All published articles of this journal are available on ScienceDirect.

Multidisciplinary Rehabilitation Approach to the Maxillo-Facial Complications of Crouson’s Disease: Case Report and Review
Authors Info & Affiliations
Abstract
Background:
Craniofacial anomalies present a challenge to all health care practitioners since they necessitate long-term team follow-up, which is difficult to achieve outside of a major center where craniofacial anomalies teams normally collaborate.
Objectives:
The current review with an illustrative case focuses on the representation and review of Crouzon syndrome and its maxillofacial implications. Review of different varieties of gene mutations that produce craniosynostosis syndromes were discussed and focused on seven clinically distinct craniosynostosis syndromes that are precipitated by the mutation in one or more of the fibroblast growth factor receptors genes which affected the maxillofacial region.
Case presentation:
A complete clinical and radiographic case scenario of a patient suffering from Crouzon syndrome was presented, and discussion of the various disciplines and techniques used along the way to achieve the best results, as well as how team collaboration and patient compliance led to the best results were represented. The presented case was treated with orthodontic treatment, Le Fort-I osteotomy, and Le Fort-III osteotomy with extraoral distraction osteogenesis.
Conclusion:
The combination of different orthognathic surgery alternatives (Le Fort-III and Le Fort-I) with distraction osteogenesis and orthodontic treatment produced excellent outcomes with few complications, and the patient was extremely satisfied and cooperative. Early and thorough team-based care for Crouzon syndrome patients should be accessible at specialized craniofacial centers.
1. INTRODUCTION
Craniosynostosis (CRS) is characterized by the early closure of cranial sutures, leading to a change in head size and shape [1]. It is not an uncommon finding and affects 1:2500 births [2]. In patients with craniofacial synostosis, the clinical presentation differs according to the sutures involved [3]. It ranges from isolated unicoronal synostosis to severe prenatal multi-sutures CRS that causes airway and feeding issues [4]. CRS could be due to external mechanical pressure, metabolic disorder, lack of underlying bone growth, or genetic mutation [1]. It could be linked to abnormalities in the skeletal, cardiac, or other organs. Many CRS syndromes are caused by several gene mutations [5]. The mutation in one or more genes of fibroblast growth factors receptors (FGFR) 1, 2, and 3 can cause seven syndromes, namely Apert, Pfeiffer, Crouzon, Jackson–Weiss, Muenke, Beare–Stevenson, Crouzon syndrome with acanthosis nigricans [6]. The most common type of synostosis is isolated sagittal synostosis, followed by coronal synostosis, with rare lambdoid and metopic synostosis [7].
Crouzon, Muenke, and Apert are characterized by a short anteroposteriorly skull (brachycephaly), and bilateral coronal CRS is one of the most prevalent abnormalities [6]. Many of the major syndromes have an autosomal dominant inheritance or are sporadic [4]. Diagnosis of these syndromes is based on clinical presentation and genetic testing [8]. Recently, 3D sonography has been found to be helpful for the early detection of morphologic anomalies, especially Apert syndrome [9]. Apert syndrome is characterized by obstructive sleep apnea, followed by Pfeiffer, Crouzon with acanthosis nigricans, and finally Crouzon syndromes with the prevalence of 80.6%, 72.7%, 66.7%, and 64.7%, respectively [10].
In Apert syndrome, the affected patient clinically has a scaphocephaly or cone-shaped head caused by early fusion of the coronal sutures, a high, prominent forehead, and a specific form of nose and mouth, such as hypoplastic midface. The term hypertelorism means an abnormally increased distance between the medial canthi of both eyes with webbing of the hands and feet [11, 12]. According to Cohen's study on seven populations, the prevalence of Apert syndrome is 15.5/1,000,000 [13]. The mortality rate of Apert syndrome is higher than in other CRS [14]. At a molecular level, the pathways that regulate proliferation, differentiation, and apoptosis of fibroblast cells are altered by the FGFR mutations. Studies have shown that pharmacological inhibitors that specifically repress these pathways could significantly improve the clinical manifestations of Apert syndrome [15]. The bicoronal synostosis is the most prevalent subtype of Apert syndrome [16]. However, congenital airway abnormalities are not common in Apert syndrome [14]. Dental manifestations of Apert syndrome include impactions, congenitally missing, severe crowding, delayed eruption, thickened gingiva, and supernumerary teeth. The maxillary arch is mostly affected, but the mandibular arch is usually involved to a lesser degree. Syndactyly is a skeletal feature of Apert syndrome that refers to soft tissue fusion of neighbouring digits in either the upper or lower extremities with or without osseous fusion [13].
Pfeiffer syndrome (PS) is an autosomal dominant disease caused by two mutations in type 1 or 2 genes of FGFR1 or FGFR2 [17]. Bicoronal craniosynostosis, acrocephalosyndactyly, middle facial hypoplasia, digital abnormalities of the hands and feet (wide thumb and large toes), and, in some cases, partial cutaneous syndactyly were the first clinical signs documented by German geneticist Rudolph Pfeiffer [18]. Cohen 1993 classified PS into three categories based on neurodevelopment and survival prognosis [19]. Type 1 has minor physical manifestations without neurodevelopmental implications and is inherited through FGFR1, whereas Type 2 is characterized by CS and hypertelorism, as well as external proptosis and a cloverleaf skull, and is caused by FGFR2 mutations [20]. Type III is the same as type II without the cloverleaf skull [20]. Type II and III are sporadic, with most patients dying early. Hearing loss is a common finding, especially in type III, due to external auditory canal and tympanic membrane aplasia [21]. Few reports are available on dental features in Pfeiffer syndrome, including hypodontia, microdontia, dilacerations, and radicular dentin dysplasia [22]. Early diagnosis of PS through prenatal ultrasound screening allows for more comprehensive care and the best possible outcomes for patients and their families.
Beare-Stevenson syndrome (BSS) is a very rare hereditary syndrome with only a few known cases worldwide [23]. It is an autosomal dominant syndrome caused by Tyr375Cys or Ser372Cys mutations in the FGFR2 [24]. Other FGFR2 mutations have also been identified [25-27]. The morphology of BSS includes (CS), particularly a cloverleaf skull, which is often apparent at birth, as well as cutis verticis gyrata, acanthosis nigricans, tags of the skin, ear abnormalities, and a prominent umbilical stump [28]. On the other hand, there were cases with all characteristics, including natal teeth and the absence of synostosis at birth [28].
A mutation in the FGFR3 gene causes Muenke syndrome, which codes for a Pro250Arg alteration [29]. This syndrome is characterized by unilateral or bicoronal suture synostosis, big toes, and carpal and tarsal fusions [30]. The phenotype ranges from no clinical manifestation to isolated synostosis to more complicated presentations overlapping other known syndromes like Crouzon, Pfeiffer, or Saethre-Chotzen [31]. Although molecular testing is required for Muenke syndrome diagnosis, macrocephaly and minor facial dysmorphisms have been found to be indirect traits [32].
The cranial deformity Crouzon syndrome (CS) is characterized by CRS, midface hypoplasia, small orbits, and ocular exophthalmos. It was described for the first time in 1912 by Octave Crouzon, which has numerous names, such as Crouzon craniofacial dysostosis or Crouzon's disease, involving both the maxilla and the mandible due to the involvement of the first branchial arch [33]. It is an autosomal dominant inherited condition mainly due to mutation related to FGFR2 [33]. The FGER2 has an important role during embryonic bone development. It represents 4.8% of all CRS with a prevalence of 1 in 25000. No evidence of sex or race predilection was reported [34]. The cranial manifestations are due to the fusion of the coronal and sagittal sutures; however, other sutures may be involved. Diagnosis of Crouzon’s syndrome is usually based on the clinical presentation, including flat sphenoid characterized bone, exophthalmos, and midface hypoplasia [35]. Suture synostosis may arise at birth or in the early years of life, causing midface/maxillary hypoplasia, shallow orbit, and occasionally upper airway obstruction [7]. Cleft lip and/or palate deformities are uncommon in individuals with Crouzon syndrome [36].
Dental features include a narrow maxillary arch, which appears as a highly arched palate, posterior dental crossbite, and crowding of maxillary teeth. 1-2 years of delay in tooth eruption is observed. Therefore, individuals with Crouzon syndrome have difficulty maintaining good oral hygiene [37]. Class III occlusion with the anterior open bite is usually present in cases with nasopharyngeal involvement/obstructions [7, 38]. Regarding ocular involvement, ocular proptosis is evident in all cases. Exotropia is reported in more than half of cases with Crouzon. Poor vision is evident in almost half cases; however, blindness is rarely reported [7].
Patients with CS with acanthosis nigricans (CANS) syndrome have a mutation in the FGFR3 gene (Ala391Glu) [14]. Due to the pleiotropy of the affected organs, CAN syndrome differs from Crouzon syndrome, and early diagnosis of CAN mutation is critical for those individuals who need to deal with affected vital organs like the kidney as soon as possible [39].
2. CASE REPORT
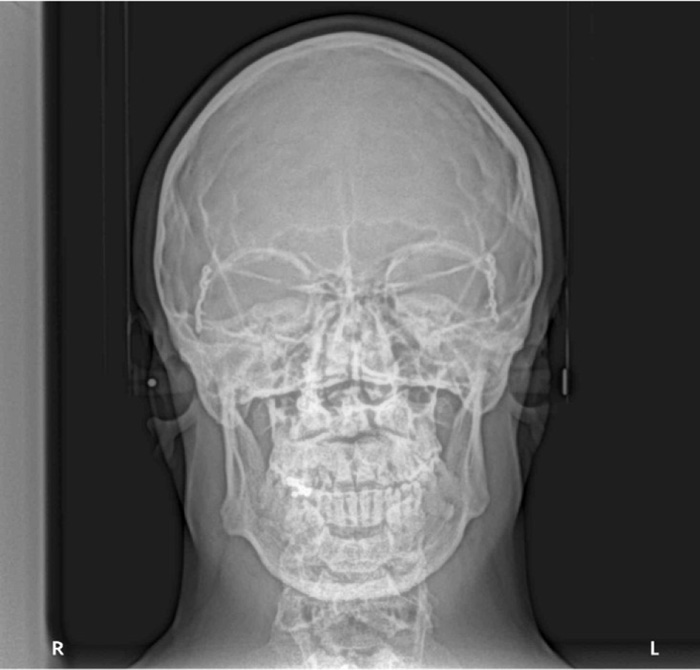
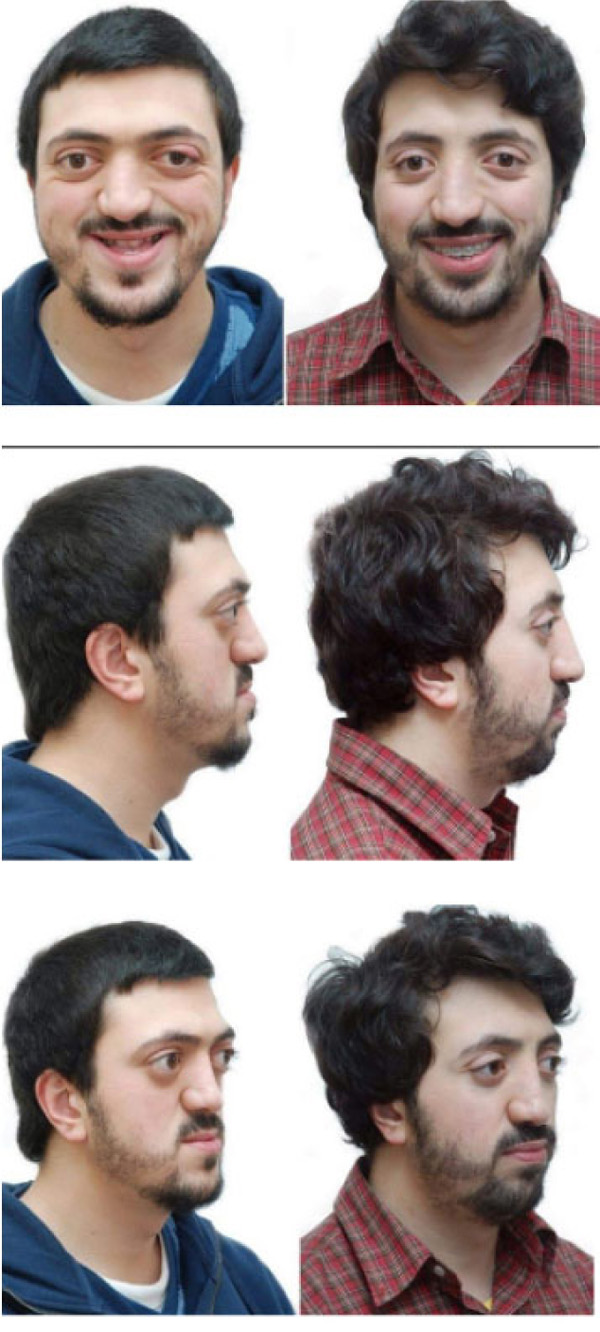
A 28 years old caucasian male was presented to the orthodontic clinic for a check-up. The patient was of medium build and showed mild frontal bossing, bilateral proptosis, large nose, hypertelorism (the distance between the medial canthus) > 31mm, mild divergent strabismus, midfacial hypoplasia, and no upper or lower extremity defects, which distinguishes CS from Apert and Pfieffer syndromes. The orthodontist diagnosed him with Crouzon’s syndrome. The patient reported that he was diagnosed with Gravis disease when he was 15 years old. In addition, he reported a history of sleep apnea and mouth breathing. The patient was mentally healthy and had no family history of a similar complaint. Intraoral examination revealed maxillary retrognathism, high narrow collapsed palate, Class-III malocclusion, crowded maxillary teeth, anterior open bite, higher buccal maxillary canines, malposed palatally erupted upper right second premolar, loss of mandibular left first molar, and malposed incompletely erupted right first premolar (Fig. 1). Radiographic examination revealed that the coronal and sagittal sutures had been obliterated and that the skull had protruding cranial marks on the inner surface of the cranial vault, which appeared as many radiolucencies looking like depressions giving the appearance of a copper hammered appearance (Fig. 2). The results of the haematological tests were normal. No genetic tests were performed.
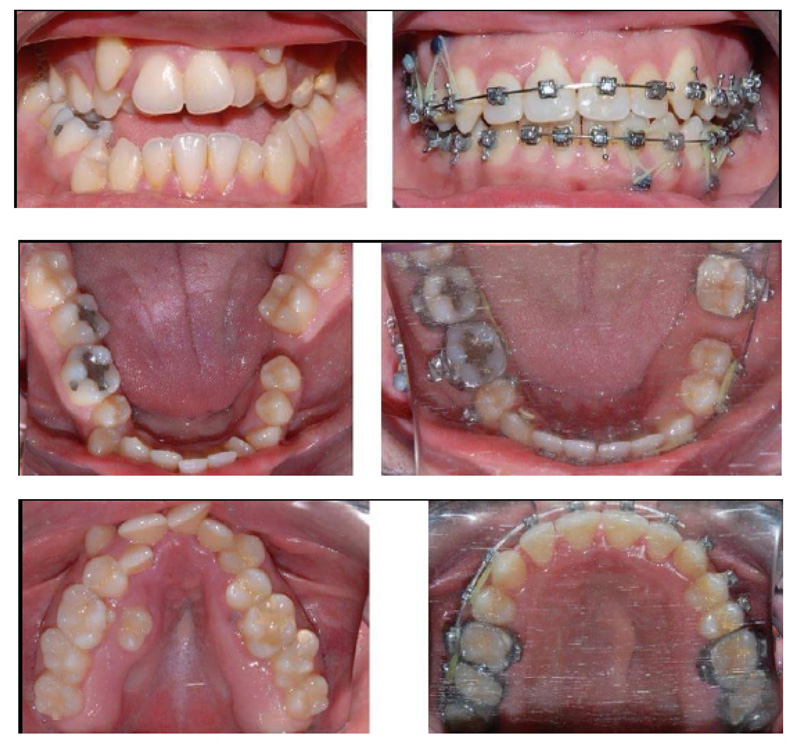
The presented case was treated with orthodontic treatment, Le Fort-I osteotomy, and Le Fort-III osteotomy with distraction osteogenesis (DO). The Le Fort-I osteotomy is a common surgery performed to correct midface deformities. It is used to treat cases with Class-II and III malocclusion, facial asymmetry, obstructive sleep apnea, and maxillary atrophy. It should be preceded by proper orthodontic treatment [40]. In this case, Le Fort-I osteotomy was performed to expand the palate. It was performed two times due to inequivalent/asymmetric expansion of the palate for the first time. Le Fort-III osteotomy was performed simultaneously with DO; it is a technique used to increase the length of the midface by moving the osteotomized bone apart, and a new bone will be formed between the separated bones [41, 42]. In this case, it was used for 11 days to protrude the maxilla (12mm) (Figs. 3 and 4). Orthodontic treatment to correct crowding and class III malocclusion started at the age of 22 and finished at the age of 26 (Fig. 1).
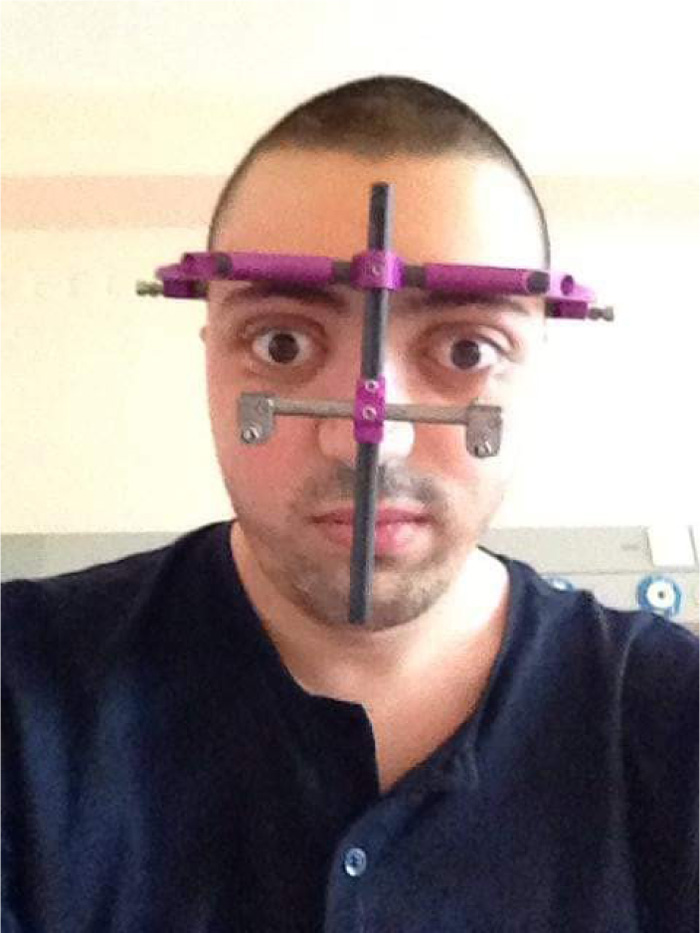
3. DISCUSSION
The purpose of this review report was to highlight an interesting case of CS that demonstrated comprehensive management of this difficult syndromic disease. In cases of Crouzon’s syndrome or other craniofacial dysostosis cases, the management is multidisciplinary, including pediatricians, craniomaxillofacial surgeons, plastic surgeons, neurosurgeons, otorhinolaryngology (ENT) specialists, ophthalmologists, orthodontists, and others according to the case to be treated.
Cranial vault remodeling is performed to increase the intracranial space and decrease intracranial pressure and the chances of an abnormal skull and upper jaw morphology during growth [43].
Cranioplasties surgical procedures are performed on children as young as 6 months of age to relieve intracranial pressure and improve the child's head appearance through open vault strip craniectomy, suturectomy, or cranial vault expansion with or without orbital advancement by a neurosurgeon. Other syndromic abnormalities (ocular, dental, ear, and respiratory) are generally treated with open surgery, which may be repeated [1, 44, 45].
Surgical options for facial rehabilitation include Le Fort-III, Le Fort-II for midfacial advancement alone or in combination with DO, Le Fort-I for maxillary advancement and palatal expansion, zygomatic repositioning and/or augmentation, monobloc and facial bipartition, and rhinoplasty, genioplasty, bone grafts as adjunctive techniques [42, 46, 47].
In the present case, Le Fort-III was used in conjunction with rigid external DO for midface advancement and achieved excellent results in improving midface features, particularly bilateral proptosis and zygomatic hypoplasia, without the need for bone grafts. On the other hand, intraoral distractors, which were not used in this case, provide less advancement and frequently require bone grafting at the end of the distraction phase [48]. This is consistent with the findings of other surgeons who reported that combining Le Fort-III osteotomy with an external distractor resulted in superior advanced outcomes with fewer problems [43, 49].
Then, Le Fort-I was required to enhance the maxillary position, conceal the false mandibular prognathism, and allow for palatal expansion in conjunction with orthodontic therapy, resulting in a very aesthetic result while closing the open bite. It was found that midface correction surgeries are better to be performed when the orbits have essentially reached physical maturity [50].
According to Mathijssen (2015), all surgeries for face correction performed after the age of 18 coincide with recommendations for orthognathic surgery [51]. The decision of what type of osteotomy to be used depends on the severity of the case, and existing anatomic considerations like exorbitism, orbital rotation, nasal length, maxillary hypoplasia, dental arch, presence of an anterior open bite, and degree of obstructive sleep apnea are all factors that need to be considered [21, 52, 53].
In cases of Crouzon's syndrome, because the maxillary jaw is frequently set back and short in all dimensions, maxillary skeletal hypoplasia leads to dental malocclusion. Orthodontic assessment should begin at a young age to avoid anterior crossbite and significant crowding of the permanent teeth, which can lead to severe skeletal imbalance [23]. When early midface progress is indicated, orthodontic therapy during mixed dentition positively impacts the occlusion [37, 43].
Following a diagnosis of any syndromic case, a thorough dysmorphology examination, genetic counselling involving craniomaxillofacial, paediatric, cognitive, audiology, dental assessment, and ophthalmology evaluations should all be included in the treatment plan.
Although the treatment of CS is a complicated process, the outcomes are promising, and an early start on this journey may save the patient from significant problems, such as blindness, hearing loss, and psychosocial trauma. Craniofacial synostosis syndromes challenge orthodontists who must work intimately with craniofacial teams to provide optimum care.
CONCLUSION
The combination of different orthognathic surgery alternatives (Le Fort-III and Le Fort-I) with DO and orthodontic treatment produced excellent outcomes with few complications, and the patient was extremely satisfied and cooperative. Early and thorough team-based care for CS patients should be accessible at specialized craniofacial centers.
AUTHOR'S CONTRIBUTIONS
HZ, HB, and SAE conceived and designed the study, conducted research, and collected and organized data. MA, GA, and AJ analyzed and interpreted data. SAE, GA, MA, and AA wrote the initial and final draft of the article. All authors gave their final approval and agreed to be accountable for all aspects of the work.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the Taibah University Ethical Committee.
HUMAN AND ANIMAL RIGHTS
No Animals were used that are the basis of this study. All the human experiments conducted according to the principles of the World Medical Declaration of Helsinki.
CONSENT FOR PUBLICATION
Participant signed a written formal consent form before surgery and consented to include the images.
STANDARD OF REPORTING
CARE Guideline were followed.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
This research did not receive specific grants from the public, commercial, or not-for-profit funding agencies.
CONFLICT OF INTEREST
The authors have no conflicts of interest relevant to this article.
ACKNOWLEDGEMENTS
Declared none.

