All published articles of this journal are available on ScienceDirect.

Effect of Nickel Chloride on Cell Proliferation
Authors Info & Affiliations
Abstract
Objective:
Metal alloys used in dentistry and in other biomedical fields may release nickel ions in the oral environment. The release of nickel might influence the normal biological and physiological processes, including tissue wound healing, cell growth and proliferation. The aim of this study was to evaluate in vitro the effects of nickel ions on cell cycle, viability and proliferation.
Materials and Methods:
Human osteosarcoma cells (U2OS) and human keratinocytes (HaCat) were exposed to different nickel chloride (NiCl2) concentrations (0 - 5mM) for various periods exposure. The viability of cultured cells was estimated by flow cytometry using Annexin V-FITC and Propidium Iodide (PI). Cell proliferation was evaluated by using carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) and flow cytometry. Finally, the effects of NiCl2 on cell cycle were assessed and quantified by flow cytometry. Statistical analysis was performed by means of ANOVA followed by Tukey’s test.
Results:
NiCl2 induced a dose and time dependent decrease in cell viability. After 24h, 1mM NiCl2 caused a similar and significant reduction of viability in U2OS and HaCat cells, while higher NiCl2 concentrations and longer exposure times showed a reduced cytotoxic effect in HaCat as compared to U2OS cells.
Exposure to NiCl2 caused a dose- and time-dependent inhibition of cell proliferation in both cell lines tested, with a prominent effect on U2OS cells. Furthermore, both cell lines exposed to NiCl2 exhibited significant changes in cell cycle distribution after 24h exposure 2mM NiCl2, as compared to untreated cells (p<0.05).
Conclusion:
Our results indicate that release of nickel ions may affect cell proliferation. The inhibition of cell growth by NiCl2 is mediated by both cell cycle arrest and by induction of cell death.
INTRODUCTION
The use of casting alloys is common place in temporary and permanent dental restorations and in orthodontic appliances because of the excellent mechanical properties [1]. Nowadays, nickel-titanium superelastic archwires are the first choice in most part of orthodontic treatments for their clinical performance [2].
The microbiological and enzymatic characteristics of the oral cavity provide a suitable environment for corrosion of casting alloys, because of physical, chemical and microbiological changes, and, even if the quality and quantity of the released cations depend upon the type of alloy and various corrosion parameters, it has been documented that some nickel-based alloys may exhibit increased corrosion, specifically at low pH [1]. Nickel ions might reach concentration levels between 10 mM and 800 mM in tissues immediately surrounding nickel alloys [3].
Studies in vitro have reported [4, 5], in simulated saliva medium, the presence of metals, such as nickel, while studies in vivo showed measurable but not toxic levels of metals in blood, saliva, and urine of patients with fixed orthodontic appliances [6-9]. Otherwise, it has been shown differences in nickel content between individuals with or without orthodontic appliances. Nickel release occurs into the dental plaque and components of saliva of orthodontic patients [10] and a significantly higher concentration of nickel can be found in oral mucosa cells of patients wearing fixed orthodontic appliances [11-13]. It has been showed that in buccal mucosal cells nickel concentrations were about 3.4-fold higher (2.52 ng/mL) than those in the control subjects [11].
Previous investigations dealing with the biocompatibility of orthodontic wires and nickel ions in vivo or utilized human or animal cells [14-18]. Nickel-containing orthodontic wires can reduce cell viability, otherwise concentrations which do not significantly modify oral epithelial cell viability and inflammatory cytokines release can stimulate apoptosis in vitro [19, 20]. Nickel ions may accumulate in human oral mucosal cells and human keratinocytes [11, 21], and may activate monocytes and epithelial cells, suppressing or promoting the expression of intracellular adhesion molecule 1 by endothelial cells, mostly depending on its concentration [22, 23]. Moreover, nickel is able to induce an up-regulation of surface antigen expression, while high concentrations may impair essential functions of macrophages stimulated by LPS [24].
Therefore, the release of nickel from biomedical and dental appliances in oral cavity might influence the normal biological and physiological process including tissue wound healing and cell growth. The aim of this investigation was to study the influence of nickel on cell proliferation in human keratinocytes and osteosarcoma cells. Human keratinocytes were used as a model of epithelial tissue cells, moreover we chose an osteoblasts cell line because it cannot be excluded that in some clinical trial, including oral surgery, osteoblasts and bone tissue may be influenced by nickel.
MATERIALS AND METHODS
Chemicals and Cell Culture
Nickel Chloride (NiCl2) and tissue culture reagents were purchased from Sigma Chemical (Milan, Italy). Human epidermal keratinocytes (HaCat) and Human osteosarcoma (U2OS) were growth respectively in McCoy's medium and in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS). Sterile stock solutions of NiCl2 were prepared directly in medium, just before use.
Cell death and Viability
Cell death and viability were quantified by flow cytometry. 7x104 cells were seeded in 35mm dishes and after 24h were treated by NiCl2 (0-5mM) for 24, 48, 72 h exposure periods. Then, floating and adherent cells were collected, washed once with PBS. Next, the cells were suspended in 500 µL of binding buffer. Untreated and treated cells were stained with annexin V-FITC and PI (MBL Medical & Biological Laboratories Co., Ltd., Nagoya, Japan), and incubated at room temperature for 15 min before being analyzed by flow cytometry (FACScan, Becton-Dickinson, San Jose, CA, USA). Viable cells (no staining), apoptotic cells (annexin V+) and necrotic cells (both PI+/annexin V+ or PI+ alone) were detected and quantified as a percentage of the entire population [25]. The sum of apoptotic and necrotic cells were considered as cell death population [26]. Analysis of the data was performed by means of the WinMDI 2.8 program. Data from at least five independent experiments were pooled to determine the mean viable cell population.
Cell Proliferation
Carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) was used to study the effect of NiCl2 on cells proliferation and growth in HaCat and U2OS cells after 24, 48, 72 h exposure periods. CFDA-SE is a fluorescein derivative which is cell permeant and non-fluorescent; cellular esterases cleave acetate groups, rendering the molecule fluorescent and cell impermeant (CFSE). Succinimidyl ester binds to free amines, resulting in long lived fluorescent adducts. CFSE is partitioned equally among daughter cells with each division, so the reduction of fluorescence intensity in the time meant cells proliferation [27]. Each cell line was harvested and washed with PBS, cells were added to equal volume of 5µM CFDA-SE and incubated at 37 °C for 10 minute with agitation. The labeling reaction was stopped for 1 min by adding an equal volume of fetal bovine serum. The CFDA-SE labeled cells were washed twice with PBS and 7x104 cells seeded in 35mm dishes. After 24h the cells were treated with NiCl2 for different incubation time. All cells were analyzed using a FACScan (Becton-Dickinson, San Jose, CA, USA). The fluorescent filters and detectors used were all standard with green fluorescence collected in the FL1 channel (530±30 nm). Samples were gated on forward scatter (FCS) versus side scatter (SSC) to exclude debris and clumps and twenty thousand events for each sample were collected. The cells were analyzed using a logarithmic amplifier to determine the percentage of stained cells and their mean fluorescence intensity. Mean fluorescence intensity of untreated cells was arbitrary assigned a percent fluorescence value of 100. Analysis of the data was performed by WinMDI 2.8 program. Each independent experiment was performed at least five times for each cell line.
Cell Cycle Analyses
Cells (5× 105) were cultured in 100-mm dishes to 70-80% confluence , then exposed to 0-5mM NiCl2 for 24 h. After each treatment, the cells were collected by centrifugation, re-suspended in 1ml cold PBS, and stored for at least 24h in 70% methanol at -20°C. Methanol was then removed after centrifugation, and the cell pellet was washed twice in PBS. Finally, the cell pellet was re-suspended in 425µl PBS, and 50µl RNAse (1mg/ml) was added for 20 min at 37°C. Then, propidium iodide (PI) (25µg/ml) was added for 5 min. The samples were analyzed with a FACScan flow cytometer (Becton-Dickinson, San Jose, CA, USA). Fluorescence signals were amplified logarithmically. Twenty thousand events for each sample were collected with the Cell Lysis software, and the data were stored as list mode files. PI signals for the analysis of the DNA content were subjected to pulse processing to assist in gating out cell debris and doublets. The percentage of cells in G1, S, and G2 phases of the cell cycle were calculated after mathematical modeling of histograms using the software Multicycle for Windows (Phoenix Flow Systems, San Diego, CA). The percentages of cell numbers in each phase were calculated from individual histograms in at least five independent experiments.
RESULTS
NiCl2 induced a dose and time-dependent reduction of viability in both cell lines tested (Fig. 1). After 24h, 1mM NiCl2 showed a significant reduction of viability in U2OS and HaCat with a decrease to about 60% (Fig 1). At higher NiCl2 concentrations (upper to 1mM) and increased time exposure, cell damage and reduction of viability was more prominent in U2OS than in HaCat cells (Fig. 1).
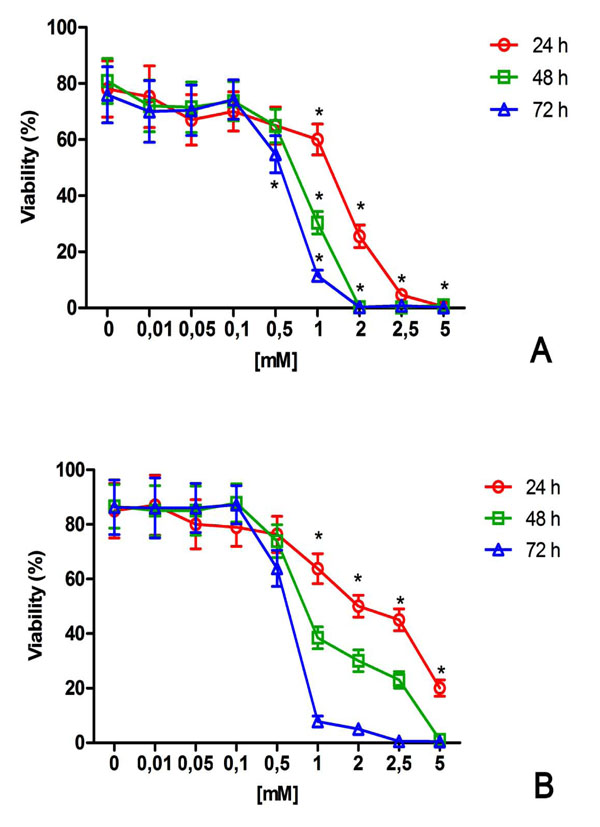
Effect of NiCl2 on U2OS (A) and HaCat (B) viability. The cells were exposed to increasing NiCl2 concentrations for 24, 48 and 72h and cell viability was detected and quantified by flow cytometry as described in the Materials and Methods (n = 5). *(p<0.05) indicates significant differences between cell culture treated with NiCl2 and untreated controls.
After 24 – 48 - 72h NiCl2 treatments, CFSE fluorescence intensity was measured to evaluate cells proliferation (Fig. 2). At 24h a 1.4 fold inhibition of cell proliferation has been show in U2OS treated with 1mM NiCl2 for 24h compared to untreated cells (Fig. 2 A). NiCl2 treatments for 48h and 72h induced a time increase inhibition of U2OS proliferation. After 72h, 0.25mM NiCl2 showed a significant cells growth inhibition reaching a maximum effect with 2 mM NiCl2 (3.4 fold) (Fig. 2A). Growth inhibition caused by NiCl2 was also found in HaCat cells (Fig. 2B). The maximum effect of NiCl2 on HaCat growth was found at 72h with a 2.3 fold inhibition caused by 2mM (Fig. 2B).
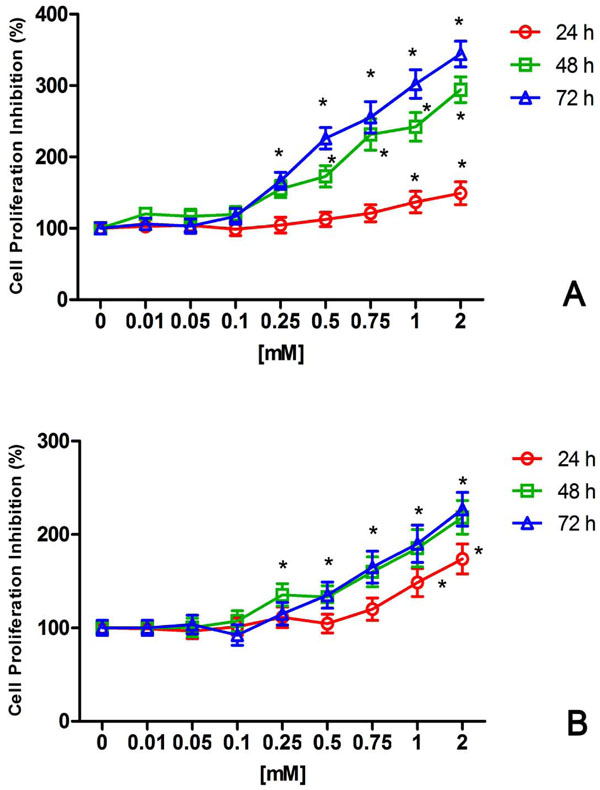
Inhibitory effect of NiCl2 on U2OS (A) and HaCat (B) cell proliferation. The cells were exposed to increasing NiCl2 concentrations for 24, 48 and 72h and CFSE positive cells were detected and quantified by flow cytometry as described in the Materials and Methods. The results were expressed and reported as the percentage of untreated growing cells (n = 5). * (p<0.05) indicates significant differences between cell culture treated with NiCl2 and untreated controls.
We also analyzed the activity of NiCl2 on the cell cycle progression of both cell lines. The distribution of cells in treated and untreated cell cultures was estimated after 24h NiCl2 exposure. U2OS exposed to NiCl2 exhibited significant changes after 24h treatments with 2mM NiCl2, while shorter time (Fig. 3A) and lower NiCl2 concentration did not significantly affect the cell cycle distribution (data not shown). The number of cells in the G1 phase was decreased from 39% in untreated control to 20% in cultures treated with 2mM NiCl2 and analyzed after a 24h incubation period (Fig. 3A). In contrast, the number of cells in S phase was significantly increased, from about 42% in untreated cultures to 58% in cultures treated with 2mM NiCl2 (Fig. 3A).
Treatments of HaCat cells with 2mM NiCl2 also induced a delay in cell cycle progression compared to untreated cells (Fig. 3B). After 24h, a significant increase of the number of cells found in G2 was observed from in cultures treated with 2mM NiCl2 (Fig. 3B).

Cell cycle analyses. The distribution of U2OS (A) and HaCat (B) cells between the three phases of the cell cycle (G1, S and G2) is presented after exposure to 1mM and 2mM NiCl2 for 24h. The percentages of cell numbers in each phase were calculated from individual histograms in at least five independent experiments (n = 5). * (p<0.05) indicates significant differences between cell culture treated with NiCl2 and untreated controls.
DISCUSSION
There is an increasing concern about the biocompatibility of dental materials and particular there is interest focused on secondary reactions caused by the use of metals. The release of nickel from dental materials has been observed in several studies [28, 29], but its toxicity remains controversial. To date, it is not known whether the release of these ions from orthodontic and dental alloys is high enough to be clinically significant [30].
Here, as previously reported [22-24], the problem of correlating in vitro cytotoxic effects to the release of nickel ions from orthodontic and dental materials was addressed by exposing cell cultures to NiCl2. In our study we evaluated the cytotoxicity grade of various concentrations of NiCl2 ranging from 0.01mM to 5mM. The NiCl2 concentrations tested here were in the range used in previous in vitro reports on other cell lines [24, 31-33]. As expected, the flow cytometry analysis (Fig. 1) revealed that the cytotoxicity increased in time and dose dependent manner. After 24h, NiCl2 exhibited cytotoxicity under a relative high concentration of 1 mM NiCl2 in both cell line tested. Interestingly, there was a different sensibility to NiCl2 between HaCat and U2OS over different time exposure and this result is in line with other studies in which is reported that the range of toxic concentration depending on cell model used [1, 18, 31-33].
So far, different in vitro methods including colorimetric, fluorimetric and radioactive, were suggested to study the effects of various dental material on cell proliferation. The most common assay to evaluate cell proliferation is based on the incorporation of radionucleotides such as tritiated thymidine (3H-TdR) into newly synthesized DNA. This method showed some limitation related to handling and disposing of radioisotopes, and to the identification only of cells which have recently synthesized DNA. In addition it does not provide information on whether labeled cells have gone on to divide or the number of cell divisions that occurred prior to exposure to the 3H-TdR pulse. Hence, several fluorescence cell-tracking dyes has been proposed to monitor cell proliferation by flow cytometry. Among fluorochromes, CFDA-SE stands out as the most versatile cell labeling dye in terms of long-term cell tracking and quantifying proliferation either in vivo or in vitro [34-36]. CFDA-SE was widely tested on various cell lines and when properly performed, the dye dilution assay correlates well with the 3H-TdR assay and other methods [37]. Here, we used CFDA-SE to analyze the effect of NiCl2 on HaCat and U2OS proliferation. Both cell line labeled showed a bright staining as reported with other cell types. The reduction of mean fluorescence intensity induced by NiCl2 clearly confirmed an inhibitory effect on cell growth in the lines tested (Fig. 2).
Finally, we examined the effects of NiCl2 on the cell cycle. The distribution of cells untreated and treated with NiCl2 was estimated after 24h exposure. Significant cell cycle changes were only detected after 24h exposure to 2mM NiCl2, while shorter time and lower concentrations did not affect the cell cycle distribution (data not shown). Interestingly, the effects of NiCl2 in the two cell lines analyzed were different. While NiCl2 induces a S-phase cell cycle arrest in U2OS cells, Hacat cells mainly accumulate in G2/M phase of cell cycle. Since p53 plays a relevant role in S phase checkpoint, it is tempting to speculate that this difference is related to the p53 status of the cells, i.e. U2OS being p53 proficient, while Hacat cells carry a mutant p53 gene. NiCl2 has recently been shown to induce inhibition of cell growth and cell cycle arrest in different cell types [38, 39] and our present data confirm and extend these data in several aspects. First, the apparent discrepancy in cell cycle responses is likely due either to cell type-specific susceptibility to NiCl2 or to differences of NiCl2 concentration used. Lower concentrations of NiCl2 (0.25 to 0.5mM) may induce G0/G1 arrest, while higher concentrations used in our study results in S and G2/M accumulation. Furthermore, we show here that, at high concentrations (0.5 to 2mM), the inhibition of cell growth by NiCl2 is mediated by both cell cycle arrest and by induction of cell death.
In conclusion, NiCl2 induces cell death and cell cycle arrest in both human keratinocytes and osteosarcoma cells. Therefore, it is plausible that NiCl2 is a candidate to affect vital processes due to an impairment of cell proliferation.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
Declared none.

