All published articles of this journal are available on ScienceDirect.

Oral Characteristics of Trisomy 8 and Monosomy 18: A Case Report
Abstract
Several reports described various mosaic chromosomal syndromes characterized by alterations originated by either an excess or deficit in the number of chromosomes. A case of mosaic trisomy 8 and monosomy 18 with significant involvement of the oral cavity is described, both in terms of general medicine and from a dental-oral perspective, and the treatment plan was planned and discussed.
Regular follow-up visits enabled to verify significant improvement in all parameters of the patient’s oral health, which urged us to press on with our quest to protect the right to health of patients affected by disabilities.
INTRODUCTION
Several mosaic chromosomal syndromes are characterized by alterations caused by either an excess or deficit of two or more chromosomes. Many anomalies have been associated with several syndromes: growth deficits, mental retardation, motor difficulties, microencephaly, agenesis of the corpus callosum, heart defects, low-set, malformed ears, hypertelorism, snub nose, and very low-set hairline. Furthermore, serious alterations have been reported in oral cavity of these patients, including cleft lip and palate, high-arched (ogival) palate, anterior open-bite and small jaw (micrognathia), and, in one reviewed case, taurodontism [1-6]. A comprehensive table of the main features (general and oral) of chromosomial anomalies is displayed in Table 1.
Main Features (General Medical and Head-Neck and Oral) of the Case Described in this Paper and Comparison of Other Chromosomial Anomalies
| Trisomy 8 Monosomy 182 | Trisomy 181-5 | Trisomy 8 Mosaicism2 | Trisomy 9 Mosaicism5 | Trisomy 20 Monosomy 113 | |
|---|---|---|---|---|---|
| General Medical features | |||||
| Malformed ears | + | + | + | + | + |
| Ocular anomalies | + | + | + | + | + |
| Hypertelorism | + | + (33% cases) | + | - | + |
| Webbed neck | + | + | - | - | - |
| Upper limbs anomalies | + | + | + | - | - |
| Lower limbs anomalies | + | + | - | + | - |
| Urogenital anomalies | + | + (33% cases) | + | - | + |
| Kidney’s anomalies | + | - | + | + | + |
| Nervous central anomalies | + | + | + | + | + |
| Head-neck Oral features | |||||
| Cleft-palate | + | - | + | - | - |
| Prognathism | + (mandibular) | + (maxillar) | + (micrognatia) | + (maxillar) | - |
| Open bite | + | - | - | - | - |
| Ankjloglossia | + | - | - | - | - |
| Narrow high-arced palate | - | - | + | + | - |
AIM
The purpose of this case-report is to describe the dental and oral situation of a young patient affected by a rare chromosomal disorder (trisomy of chromosome 8 and monosomy of chromosome 18 mosaic) previously unreported in literature and to illustrate the treatment plan.
Case Description
The patient, male aged 12-year-olds, arrived at the Dental Clinic of the University of Sassari taken by his mother. She reported that, the children experienced discomfort when ingesting food and liquids, and that he had several caries lesions.
The medical history of the patients pointed out a complex clinical picture. Moreover, the young patient was completely uncooperative and persisted on keeping his mp3 headphones on; his mother indicated this was the only way to “keep him quiet and calm”.
General Medical History
The medical history together the documents provided by the mother indicated that the patient is the third-born child in his family, with two older brothers. Both parents and siblings are in apparent good health. The child was conceived when his father was 31 and his mother 24. In consideration of his malformations, previously diagnosed via ultrasound scan a Caesarean-section was performed at 40 weeks gestation.
The mother’s pregnancy had presented no noteworthy complications. At birth the patient showed: dysmorphic facies with brachycephaly, low forehead with low-set hairline on the forehead, short eyelid rims with slightly mongoloid attitude, esotropia, and hypertelorism. Furthermore, in the patient were observed evident manifestations of unilateral complete cleft of left primary and secondary palate, ipo-maxilla, low-set ears with flattened and slightly pointed helixes, prominent antitragi with wide and stubby lobules and short and webbed neck. Several other anomalies were also present in both upper and lower limbs and in the external genitalia.
Echoencephalogram revealed agenesis of the corpus callosum with colpocephaly and ultrasound scan of the kidneys evidenced first degree, right side pyelectasis.
At the age of two, the patient was referred for genetic analysis (Regional Center for the Ente Ospedaliero Ospedali Galliera in Genoa), where an in-situ hybridization test was performed on lymphocyte metaphases from peripheral blood.
The results evidenced a two cell-line karyotype with reciprocal translocation between a chromosome 8 and a chromosome 18:
(46,xy,t(8;18)(8qter→8p21.1::18q23→18pter→18q23::8p21→8pter//.46,xy,t(8;18)(8qter→8p21.1::18q23::18qter→18q23::8p21.1→8qter; 8pter→18q23::8p21.1→8pter.
It was not until 2000, that the presence of transmissive hypoacusia and epilepsy were also revealed, as well as subvalvular aortic stenosis (corrective surgery was performed in 2001), gastroesophageal reflux, and severe psico-motor retardation.
Oral Examination
The complete lack of patient’s cooperation made the intra-oral examination extremely tough to perform. An examination of the patient’s face evidenced prognathism of the lower jaw, labial incompetence with sequela of cleft upper lip cicatrisation, a significant anterior open-bite, hypertelorism, snub nose, low-set hairline, low-set ears with flattened and slightly pointed helixes, and prominent antitragi with wide and stubby lobules (Figs. 1-2).
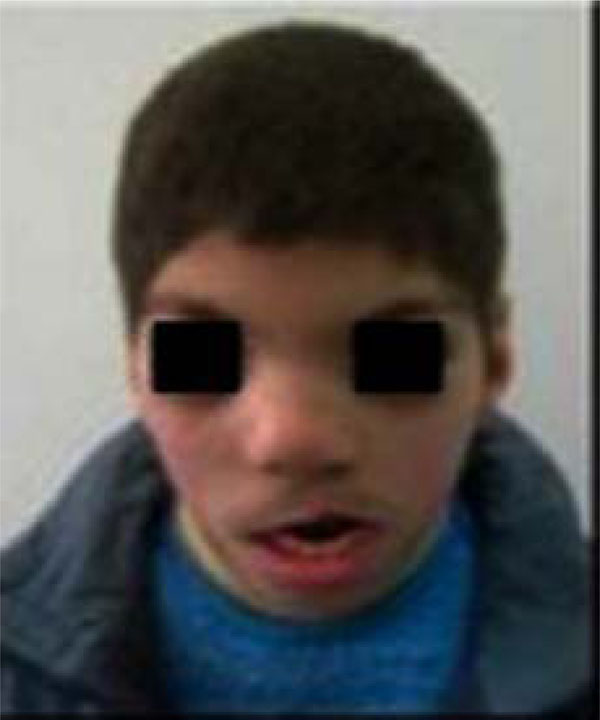
Frontal view of the patient.
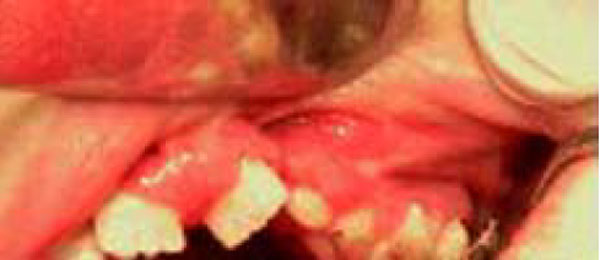
Sequela of cleft upper lip cicatrisation.
It was observed a severe shortened lingual frenulum leading to ankyloglossia (probable cause of the jaw’s appearance), and cleft soft palate with disruption of the alveolar process. Moreover, a great amount of plaque and calculus was observed on the teeth with a severe gingivities, as well cavities on elements 16 and 36, with the following dental configuration: 16, 15, 14, 13, 12, 11, 21, 23, 64, 65, 26, 36, 75, 74, 73, 32, 31, 41, 42, 43, 84, 85, 46 with persisting of several deciduous elements in the 2nd, 3rd, and 4thquadrants (64, 65, 73, 74, 75, 84, 85) and agenesis of element 22.
Description of Intervention
After undergoing a cardiologic visit and being examined by anesthesiologists, the patient was admitted to Day Surgery 7.30 am. After that, the patient was intubated and placed under anaesthesia professional oral cleaning was performed (Fig. 3). This procedure further highlighted the presence of cavities and the persistence of numerous deciduous elements in the dental arch. An intervention geared towards preserving elements 16 and 36 was conducted using reconstructive silver amalgams. Finally, a lingual frenulectomy (Fig. 4) was performed, and elements 64, 65, 73, 74, 75, 84, and 85 were removed.
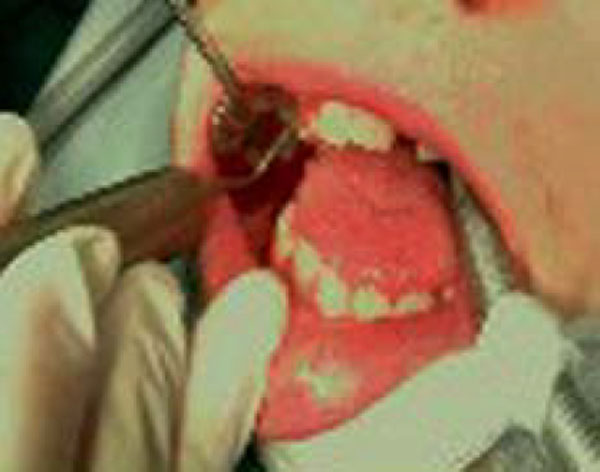
Operative treatment. Professional oral hygiene.
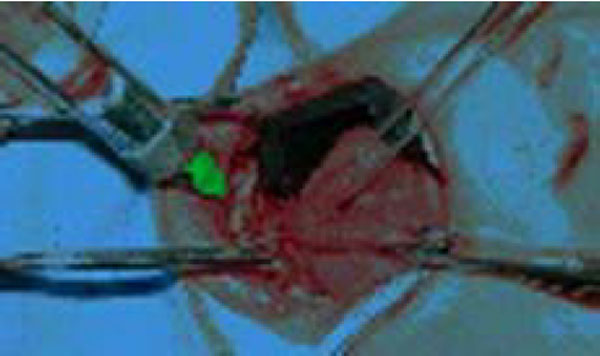
Lingual Frenulectomy.
Release from Hospital
After regaining consciousness without complications, the patient remained under observation as needed and was released later in the afternoon of the same day. Before the patient was released, his parents received basic instructions to be followed during the first post-surgery days (wide-spectrum antibiotic and anti-inflammatory treatment and food intake limited to cold liquids). A follow-up visit was set up for ten days later.
DISCUSSION AND CONCLUSIONS
To the best of the authors’ knowledge this is the first report describing the oral characteristics of a rare chromosomal disorder (trisomy of chromosome 8 and monosomy of chromosome 18 mosaic). The case described is characterized by several manifestations previously reported on in the literature e.g. the trisomy of chromosome 18 and 2, trisomy 8 mosaicism, Jacobsen syndrome 1, such as clubfoot dimorphism of the ears, heart disease, abnormal facial features and seriously altered stomatognathic system. As described in Table 1, previously unreported features included prognathism of the macrognatic lower jaw, ankyloglossia and hypodontia that should be expression of this complex chromosomal disorder. These anomalies and dimorphisms make the clinical picture extremely complicated, rendering the case approach quite challenging both in terms of systemic concerns and oral cavity related issues.
Our experience and intervention, albeit belated, enabled us to observe a distinct improvement in the patient’s oral health during the three months of follow-up visits, with an improved oral hygiene at home; moreover no new cavities were observed. Finally, the psychological impact for the parents should not be underestimated, they finally felt directly involved in maintaining their child’s oral and nutritional health. This decreased his discomfort despite his serious medical condition.
Often oral health is underestimated in patients affected by serious systemic disorders; this can lead to dramatic clinical situations with regard to cavities and periodontal disease, sometimes leading to loss of teeth even in young patients.
Improving the disabled patient’s oral health should instead be viewed as a right and not a privilege. In fact, disabled people have a right to a health standard comparable with that afforded to other patients.

