All published articles of this journal are available on ScienceDirect.

Oral Surgical and Haematological Management in a Female Patient with Turner Syndrome and Moderate Haemophilia A: Clinical Observation and Case Report
Authors Info & Affiliations
Abstract
Introduction:
Turner syndrome patients are at higher risk of having X-linked recessive disorders that could have serious clinical implications. Somatic abnormalities that may coexist with coagulation disorders determine the medical procedure approaches.
Case Report:
We report a 29-year-old female showing dysmorphia, distinctive physical features, and coagulation disorder, referred for maxillofacial surgery. Based on clinical symptoms, the patient was diagnosed with Turner Syndrome, and haemophilia A. Karyotyping confirmed classical monosomy X in all analysed blood cells. Molecular studies revealed hemizygous point mutation c.5096A>G (p.Tyr1699Cys) in Factor VIII gene, in exon 14. This missense mutation disturbs the interaction of Factor VIII with the von Willebrand factor, causing moderate haemophilia in the proband. The article presents the clinical history and preparation of our patient for oral surgical and dental surgery treatment.
Conclusion:
Turner syndrome patients require special attention due to the higher probability of congenital haemorrhagic diathesis. Maxillofacial surgery interventions in Turner syndrome and congenital haemorrhagic diathesis patients require individual patient preparation preventing post-extraction bleeding and ensuring proper local haemostasis.
1. INTRODUCTION
The congenital developmental disorder known as Turner Syndrome (TS) is characterised by a series of phenotypic abnormalities. Turner syndrome was described in 1938 by Turner. It concerned the description of a seven-year-old girl distinguished by short stature, a webbed neck, elbow deformity, and underdevelopment of secondary sex features. Sex chromosome disturbances determine observed somatic features [1-4].
The incidence of TS was estimated from 1: 2000 to 1: 2500 female newborns. Studies conducted by Fordin in 1959 showed a lack of one X chromosome in all or some cells. Genetic testing confirmed that 40-60% of TS cases have a 45,X karyotype called X monosomy. Less frequent structural X chromosome aberrations are observed in 12-20% of TS patients. So-called mosaic karyotypes occur in about 30-40% of women with TS [1, 2].
In addition to the above frequently described somatic anomalies, including short stature and stocky body, many other abnormalities have been distinguished in TS cases, e.g., in the masticatory organ, eyes, hearing, and internal organs (kidney hypoplasia, abnormalities in the urinary and cardiovascular systems). Diabetes and thyroid function abnormalities are also common disorders associated with TS [5, 6].
The specialist literature contains single case reports of TS symptoms coexisting with coagulation disorders, manifested as spontaneous and/or prolonged bleeding [7-9]. Shahriari et al. in 2016 described the case of a 6-month-old girl with prolonged postoperative bleeding related to ophthalmological surgery [9]. The child's blood screening parameters were normal; however, extended tests revealed abnormalities in factor VIII (FVIII) plasma activity. Genetic diagnostics tests detected a mosaic type of Turner syndrome [9].
Delayed diagnosis of coagulation factor deficiencies could have serious clinical implications [7]. Patients with TS are at higher risk of having X-linked recessive disorders due to monosomy. Mutations of genes coding factor VIII (F8) and IX (F9) on the X chromosome are inherited or could appear as de novo and are responsible for coagulation defects in female patients with TS [8, 10, 11].
In clinical practice with TS patients, we usually concentrate on somatic abnormalities associated with monosomy, but we should also focus our attention on other symptoms or abnormalities that could be caused by other coexisting genetic abnormalities [10, 12-14].
2. THE AIM
The report aims to present a rare case of female patient with Turner syndrome and a moderate haemophilia A in whom dental extraction was performed, obtaining stable local haemostasis and preventing secondary bleeding after tooth extraction.
3. CASE PRESENTATION
A 29-year-old female patient with odontogenic inflammatory infiltration and an oral abscess was admitted to our clinic for treatment due to the previously diagnosed coagulation disorder. The patient experienced inflammation in the form of dental pulp necrosis of teeth 17 and 24. Due to this, she was prescribed antibiotics and painkillers. In the past, she underwent endodontic treatment. However, her teeth were not eligible for the conservative and endodontic treatment approach, and it was the indication for their surgical removal. The patient declined the extraction of these teeth formerly. Consequently, the crown of tooth 24 broke off at the level of the tooth's neck. Surgical removal of tooth 17 and tooth-root 24 was undertaken in our clinic.
At the age of 8, the patient was diagnosed with moderate haemophilia A when following tonsillectomy, she experienced heavy, difficult to control bleeding. Factor VIII activity in plasma (one-stage assay) was 3 IU/dl (normal range 50-150 IU/dl). The patient declared that in the last years, there were no spontaneous or traumatic bleeding episodes that would require substitution treatment.
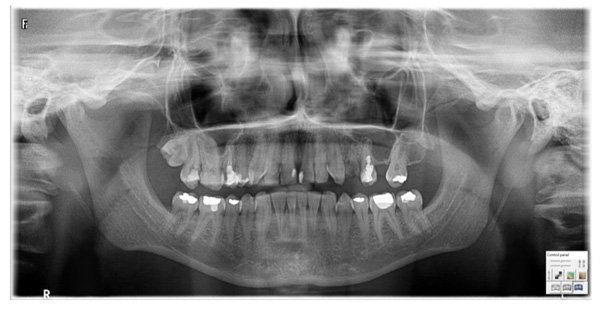
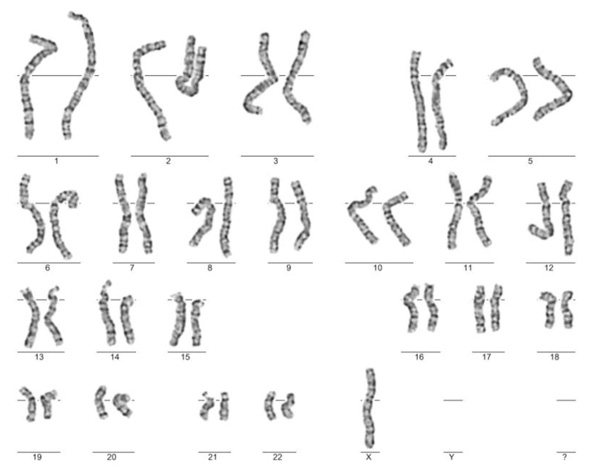
According to the clinical interview, Turner syndrome was diagnosed at six years old, genetic testing confirmed karyotype 45,X (Fig. 1). For the last ten years, the patient was receiving levothyroxine sodium for hypothyroidism in the course of Hashimoto disease. Due to heavy menstrual bleeding, the patient received tranexamic acid; she has no menstruation currently.
The patient's phenotype is typical for individuals with Turner syndrome, short stature, height 139 cm, body weight 76 kg, wide neck, short chest, poorly developed breasts, nipples wide apart, low descending hairline, elbow deformity (cubitus valgus). The patient had hypotonia of mimic muscles causing facial dysmorphia. Body skin was clean without pathological patches or discolouration. Osteopenia of the skeletal system was recognised. Blood pressure (on the day of admission) was 160/95 mmHg.
Mouth and local condition examination revealed the typical shape of the lips and neglected oral hygiene. Bite disorder in which upper front teeth almost completely overlap the lower front teeth (deep – overbite) was indicated. It was noticed that the vestibule of the mouth is flattened. The numerous missing teeth in the area of the upper dental arch were detected. Crowns on teeth 17, 24 were damaged by the process of caries and dental pulp necrosis. The mucosa of the part of the vestibule oral cavity and the floor of the mouth and hard and soft palate were normal. A large tongue without any pathological changes of the surface, highly arched hard palate, and soft palate with normal construction were observed.
The results of blood tests are listed in Table 1.
| Coagulation Parameters | Haematological Parameters | Biochemical Parameters | Hormones | ||||
|---|---|---|---|---|---|---|---|
| Coagulation Factor VIII Activity [%] | 3 (>50) | WBC [x103/µl] | 7,04 (4-10) | Glucose [mmol/L] | 4.94 (3.4-5.5) | TSH [µlU/ml] | 2,55 (0.27-4.2) |
| PT [s] | 8.9 (10-13) | RBC [x106/µl] | 5,14 (3.5-5.0) | Bilirubine [µmol/L] | 13 (0-17.1) | FT4 [ng/dl] | 1.17 (0.9-1.9) |
| Fibrynogen [g/L] | 2.8 (2-4) | HGB [g/l] | 14,8 (11-15) | Urea [mmol/L] | 2.3 (1.7-8.3) | FSH [mlU/ml] | 4.43 |
| APTT [s] | 44.7 (26-36) | HCT [%] | 44,1 (37-47) | Creatinine [µmol/L] | 56.6 (45-97) | LH [mlU/ml] | 2.99 |
| PLT [x103/µl] | 321 (125-340) | ALT [U/L] | 19 (10-31) | Estradiol [pg/ml] | 246.0 | ||
| NEU [x103/µl] | 4,71 (2.4-7.0) | AST [U/L] | 22 (10-31) | ||||
| Na [mmol/L] | 138 (138-147) | ||||||
| K [mmol/L] | 4.41 (3.5-5.5) | ||||||
3.1. Orthopantomogram
The orthopantomogram showed the following results: Normal bone structure, trabecular form preserved. Tooth necrosis 17. Chronic inflammation of the periapical tissues and in the area of the tooth root 24. Lesions proximal to the maxillary sinus (Fig. 1).
3.2. Genetic Testing
Chromosomal GTG banding was performed on metaphases from blood in accordance with standard procedures. Karyotyping revealed monosomy of the X chromosome in all analysed cells (Fig. 2). Molecular testing was carried out on genomic DNA extracted from peripheral blood leukocytes using the standard salting-out method. Screening for inversions in the F8 intron 22 (INV22) and intron 1 (INV1) was performed using inverse shifting-PCR (IS-PCR) as previously described [15]. Bidirectional sequencing analysis of all coding regions and intron/exon boundaries of the F8 gene was conducted using the Sanger method (Genetic Analyser Abi Prism 3130XL). Sequence Variants have been reported according to HGVS Nomenclature.
The presence of inversions in intron one and intron 22 was excluded. Sequence analysis of the F8 gene revealed a hemizygous missense variant c.5096A>G (p.Tyr1699Cys) (Legacy AA No. 1680) in exon 14. This sequence variation has been reported in the locus-specific F8 mutation databases: Factor VIII Variant Database (http://www.f8-db.eahad.org/), CDC Hemophilia A Mutation Project (CHAMP; https://www.cdc.gov/ncbddd/hemophilia/champs.html) and The Human Gene Mutation Database (CM900091; http://www.hgmd.cf.ac.uk/) in association with mild or moderate haemophilia A. Sulphation of Tyr1699 (previous legacy Tyr1680) of factor VIII has been reported as essential for the interaction of factor VIII with von Willebrand factor (VWF) [16]. Substitution of tyrosine with cysteine (p.Tyr1699Cys) probably results in impaired VWF binding in the a3 domain of the factor VIII light chain, which is crucial for the survival of FVIII in patient plasma.
3.3. Surgical Treatment
Based on the clinical examination and medical documentation, the patient qualified for incision of the intraoral abscess and removal of the tooth root 24 as sanitation of the oral cavity. In preparation for the procedure, about 30 minutes before the surgery, desmopressin (0.3 µg/kg body weight) and tranexamic acid (3 x 1 g) were administered intravenously. The wounds after removal of the teeth were treated locally and carefully using haemostatic fibrin-coated collagen fleece (TachoComb®).
Under general anaesthesia, the abscess was incised and drained in the area of 15-17. Tooth 17 and tooth-root 24 were removed. During root extraction of tooth 24, the connection with the maxillary sinus was found. The wound and the junction with the maxillary sinus in the area of 24 were covered with a preparation of TachoComb forming a mucoperiosteal plate according to the Wassmund-Borusiewicz technique (the Wassmund procedure, modified in 1948 by Borusiewicz, which recommends the extension of the vestibular flap by cutting the periosteum at its base [17]). The postoperative course was uneventful.
Haemostasis was achieved after surgery. Intravenous administration of desmopressin together with the use of tranexamic acid and local haemostatic fibrin-coated collagen fleece ensured a stable clot and normal local haemostasis.
In the postoperative period, the patient received tranexamic acid 1 g 3 times a day in intravenous infusion, and subsequently, tranexamic acid was orally administered.
The outcome of the treatment was evaluated on the basis of secondary bleeding after tooth extraction. The teeth were healing properly. Minor bleeding from tooth socket 17 occurred on the third day after surgery, which resolved after applying pressure and administering 1 g tranexamic acid as an intravenous infusion. On the fourth day after surgery, the patient was discharged for outpatient treatment with the recommendation of taking tranexamic acid three times a day with a dosage of 1 g until extraction wounds have been healed. The patient has reported a follow-up visit seven days after surgery. There were no signs of bleeding. The wounds healed properly without inflammation.
4. DISCUSSION
Turner syndrome is associated with well-known somatic symptoms, which, once clinically and genetically diagnosed, do not cause major therapeutic issues in clinical practice [1, 2, 4, 5]. The patient described in the present paper is an example of a case with other comorbidities. In many medical specialities, particularly surgery, patients with TS require special attention due to the possibility of haemorrhagic diathesis occurrence [7, 9]. The specialist literature available contains individual reports of Turner syndrome cases with symptoms manifested by spontaneous bleeding [7-9]. The cases described so far concerned general surgical procedures, not encountered papers regarding oral bleeding after oral surgical and dental procedures.
In the last decade, the wide availability of new fibrin-collagen patches used for local haemostasis in patients, among others with congenital coagulation disorders, enables individual planning of dental surgical procedures in selected cases. This effective tool was used in the presented case of moderate haemophilia A. In the preparatory for an oral surgical procedure, desmopressin was adopted due to the asymptomatic long-term course of haemorrhagic diathesis, considering this approach individual, as described elsewhere e [18, 19].
The clinical observations presented in patients with Turner syndrome and coexisting moderate haemophilia A, in which desmopressin and tranexamic acid and local protection of the post-extraction wound with a collagen-fibrin patch instead of FVII supplementation were used, confirmed the safety of the applied therapeutic approach in this case. The above procedure, undertaken individually in our patient, protected against the occurrence of prolonged and secondary post-extraction bleeding and ensured proper local haemostasis.
CONCLUSION
The chromosomal abnormalities found in X monosomy result in a distinctive appearance of patient phenotype. However, they may additionally coexist with other genetic abnormalities, mainly recessive X-linked disorders, as seen in the above-described patient with haemophilia A. Therefore, during the preparation of patients for surgery, all medical specialities should pay special attention to female patients with Turner syndrome who have a higher probability of congenital haemorrhagic diathesis.
AUTHOR CONTRIBUTIONS
BL and RB collected clinical information and drafted the manuscript.
AM performed genetic testing and drafted the manuscript.
EO and JW contributed to genetic testing.
All authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patient’s parents to publish medical records.
STANDARDS FOR REPORTING
CARE guidelines and methodology were followed.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We are grateful to the patient and her parents for their cooperation.

