All published articles of this journal are available on ScienceDirect.

Systemic Chemical Desensitization of Peptidergic Sensory Neurons with Resiniferatoxin Inhibits Experimental Periodontitis
Authors Info & Affiliations
Abstract
Background and objective:
The immune system is an important player in the pathophysiology of periodontitis. The brain controls immune responses via neural and hormonal pathways, and brain-neuro-endocrine dysregulation may be a central determinant for pathogenesis. Our current knowledge also emphasizes the central role of sensory nerves. In line with this, we wanted to investigate how desensitization of peptidergic sensory neurons influences the progression of ligature-induced periodontitis, and, furthermore, how selected cytokine and stress hormone responses to Gram-negative bacterial lipopolysaccharide (LPS) stimulation are affected.
Material and methods:
Resiniferatoxin (RTX; 50 μg/kg) or vehicle was injected subcutaneously on days 1, 2, and 3 in stress high responding and periodontitis-susceptible Fischer 344 rats. Periodontitis was induced 2 days thereafter. Progression of the disease was assessed after the ligatures had been in place for 20 days. Two h before decapitation all rats received LPS (150 μg/kg i.p.) to induce a robust immune and stress response.
Results:
Desensitization with RTX significantly reduced bone loss as measured by digital X-rays. LPS provoked a significantly higher increase in serum levels of the pro-inflammatory cytokine tumour necrosis factor (TNF)-α, but lower serum levels of the anti-inflammatory cytokine interleukin (IL)-10 and the stress hormone corticosterone.
Conclusions:
In this model RTX-induced chemical desensitization of sensory peptidergic neurons attenuated ligature-induced periodontitis and promoted a shift towards stronger pro-inflammatory cytokine and weaker stress hormone responses to LPS. The results may partly be explained by the attenuated transmission of immuno-inflammatory signals to the brain. In turn, this may weaken the anti-inflammatory brain-derived pathways.
INTRODUCTION
Understanding the complex pathophysiology of periodontitis continues to be a major challenge in clinical and experimental research. Recognition of microorganisms and subsequent activation of innate and adaptive components of the immune system are essential for resistance to infectious and inflammatory diseases [1]. Sensory peptidergic nerves are major regulators of immune/inflammatory reactions, and the number of identified bioactive neuropeptides is ever-increasing [2, 3]. Via neural and hormonal pathways the brain controls and regulates many of these immune responses, and dysregulation of the overarching regulatory pathways may play a significant role for susceptibility/resistance to infectious and inflammatory diseases [4-12].
Cells of the innate immune system express pattern recognition receptors (PRRs) that detect invariant molecular structures on microorganisms, including Toll-like receptors 4 (TLR4) that sense lipopolysaccharide (LPS) on Gram-negative bacteria [13]. When such pathogen-associated molecular patterns (PAMPs) are perceived, distinct intracellular pathways are activated. Expression of a wide variety of genes ultimately results in production and secretion of immune mediators, including pro- and anti-inflammatory cytokines. These and a number of other immune mediators are of critical importance in co-ordinating innate and adaptive immunity, whose cooperative interactions enable the immune system to recognize, eliminate, and control pathogens with maximal efficacy and minimal damage to surrounding tissues [13]. In addition, LPS is directly and indirectly (via immune mediators) sensed by terminals of a subpopulation of primary afferent sensory nerves (capsaicin-sensitive nerves) that release several neuropeptides with immunoregulatory properties, such as substance P, calcitonin gene-related peptide, and somatostatin, from peripheral nerve terminals at the site of inflammation [14], as well as transmit information to the brain [15, 16].
Stimulation of the sensory nerves with LPS or immune mediators is, among others, conveyed to the parvocellular neurons within the paraventicular nucleus of the hypothalamus and neurons in the locus coeruleus, which in turn activates the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) [3, 11]. The subsequent release of glucocorticoid hormones and catecholamines regulate a number of immune system responses whose main effect is to down-regulate pro-inflammatory and up-regulate anti-inflammatory responses [3, 7, 8, 10]. LPS may also act upon the parasympathetic nervous system (PSNS). Like the HPA axis and SNS it has anti-inflammatory properties, mostly due to liberated acetylcholine [7]. These brain-controlled regulatory pathways strive to skew immune responses towards the anti-inflammatory side and thus importantly determines the natural course of infectious and inflammatory diseases.
Our previous research has revealed that the reactivity of the HPA axis, SNS and PSNS vitally influences the susceptibility and resistance to periodontitis [17-27]. Until now it has not been demonstrated if and how the sensory peptidergic nervous system can modulate the clinical course of periodontitis. Due to the effect on the non-selective cation channel “transient receptor potential vanilloid 1” (TRPV1), high systemic doses of capsaicin, or the much more potent capsaicin analogue resiniferatoxin (RTX), reduce or abolish the sensitivity of these nerves in rodents [15, 28-31]. Since LPS and LPS-induced immune mediators stimulate peripheral sensory peptidergic nerve terminals to release neuropeptides at the site of inflammation, and peripheral sensory nerve activation generates signals to the brain and activates efferent anti-inflammatory pathways, experimental sensory peptidergic desensitization may also be used to examine how the sensory peptidergic nervous system reacts to exogenous stimulants like LPS to modulate the disease process. To potentially elucidate mechanisms, we have included measurement of selected pro- and anti-inflammatory cytokines as well as the HPA axis-derived hormone corticosterone after a systemic challenge with LPS.
MATERIAL AND METHODS
Animals
Twenty male Fischer 344 rats, weighing 250-260 g, were obtained from Möllegaard Breeding Center (Ejby, Denmark) and used after 2 weeks of acclimatisation. Fischer 344 rats were used because our previous experiments have revealed that compared to histocompatible and stress low responding Lewis rats, these stress high responding animals are highly susceptible to periodontitis [17, 18, 32]. Standard rat chow pellets and tap water were available ad libitum. The animals were housed in groups of five under a 12/24 h light/dark cycle (light on from 7.00 a.m. to 7.00 p.m.) with temperature and humidity at 22 °C and 40-60 %, respectively. The experiments were registered and approved by the Norwegian Experimental Animal Board (NEAB).
Chemical Desensitization of Sensory Peptidergic Neurons with Resiniferatoxin
Systemic chemical desensitization of sensory peptidergic neurons was induced with RTX 50 μg/kg (Sigma-Aldrich, St. Louis, MO, USA,) [14]. RTX was dissolved in absolute ethanol to make a 1 mg/ml stock solution and was further diluted with saline. One group of animals (n = 10) was injected subcutaneously with this solution once daily on three consecutive days in doses of 50 μg/kg body weight. The controls (n = 10) received vehicle (ethanol diluted with saline) only. The success of RTX treatment was controlled by the “wiping test” [14]. In short, 100 µg/ml 100 µl of capsaicin solution was placed onto the cornea and the number of eye wipes was counted for 1 min. None of the desensitized animals showed wiping behaviour.
Experimental Periodontal Disease
Two days after the last injection of RTX, all animals were anaesthetised with a subcutaneous injection of Hypnorm-Dormicum (fentanyl/fluanizone, midazolam; Janssen and Cilag, Saunderton, U.K.) 0.2 ml/100g body weight. A sterile silk ligature (Ethicon Perma-hand® size 3/0, Norderstedt, Germany) was tied around the neck of the maxillary right 2nd molar tooth in the gingival sulcus. The ligatures served as a retention device for oral microorganisms. Twenty days after application of the ligatures, all animals were killed by decapitation. The maxillae were excised and fixed in 4 % formaldehyde.
Lipopolysaccaride Challenge
To assess whether the treatment regimen influenced cytokine or corticosterone responses, all animals were injected with LPS (E.coli serotype 0111:B4, Sigma-Aldrich, St. Louis, MO, USA) 150 μg/kg intraperitoneally 2 h before ending the experiments. After decapitation of the rats, blood samples were collected (6 - 10 ml from each animal) in vacutainer tubes (10 ml without additives) and allowed to clot on ice for 1 h. Thereafter, the samples were centrifuged for 20 min at 2000 x g. The serum samples were removed, aliquoted and stored at –20 ºC prior to analysis.
Assay of Serum TNF-α , IL-10, and Transforming Growth Factor (TGF)-1β
The concentrations of TNF-α , TGF-1β, and IL-10 were measured by means of enzyme-linked immunosorbant assays (ELISA) kits (R&D systems, Inc., Minneapolis, MN, USA) with catalogue numbers RAT00 for TNF-α , MB100 for TGF-1β, and R1000 for IL-10. The minimum detectable concentration for TNF-α was less than 12.5 pg/ml, and less than 31.2 pg/ml for TGF-1β and IL-10.
Corticosterone Assay
Corticosterone was measured with 125I radioimmunoassay (RIA) coat-A-count kit from Diagnostic Products Corporation, Los Angeles, CA, USA, catalogue number TKRC1. The detection limit was 5.7 ng/ml.
Radiographic Examination
The specimens were stabilised with dental wax on a Sidexis digital X-ray sensor, orientated with the axis of the teeth parallel to the sensor surface by using 4x magnification loupe glasses (Zeiss, Norstedt, Germany). The distance between the cemento-enamel junction and bone on mesial surfaces of the 2nd molars was displayed digitally. The examination was done blinded. Each X-ray was read three times, and the mean of the three readings calculated. The reliability of the method has been tested earlier [17]. Bone loss as measured with digital X-rays was chosen as an indicator of the severity of periodontitis because our previous studies have shown that this measurement is significantly more reliable than measuring periodontal fibre loss and bone loss on histological sections [17-22].
RESULTS
Effect of Resiniferatoxin Pre-Treatment on the Weight of the Animals
The RTX-treated and control animals weighed 254.1 ± 7.6 g and 252.9 ± 8.5 g (p = 0.69), respectively, at RTX induction, and 281.0 ± 12.3 g and 304.2 ± 11.4 g (p < 0.01), respectively, at the end of the experiments.
Effect of resiniferatoxin pre-treatment on periodontal tissue destruction
The RTX-treated animals had significantly less alveolar bone loss than the controls (615 ± 22 µm vs. 700 ± 15 µm; p < 0.01; Fig. 1). Since the RTX-treated rats weighed less, the length of their teeth could be shorter. We therefore compared the root-length of the right 2nd molar teeth in the two groups by measuring the distance between the cemento-enamel junction (CEJ) and apex on mesial root surfaces. There was no difference between the root-length in the RTX- and saline- treated control rats.
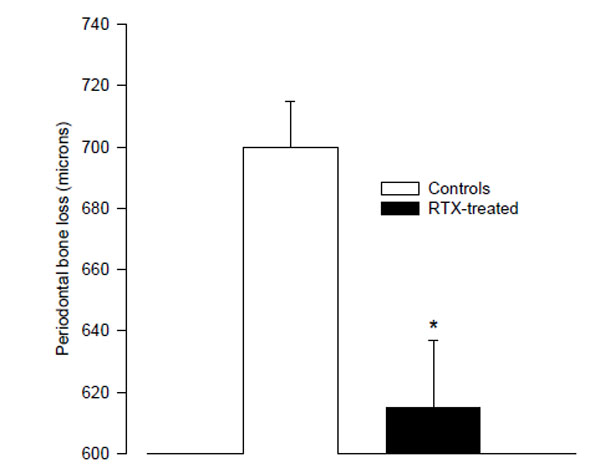
The mean distance from the cemento-enamel junction to the alveolar bone crest in resiniferatoxin-treated rats, and vehicle-treated control rats as measured on digital radiographs. (* p < 0.01 vs. controls).
Effects of Resiniferatoxin Pre-Treatment on Selected Serum Cytokines after LPS Challenge
After LPS challenge RTX-treated rats tended to react with higher TNF-α serum levels than vehicle-treated control rats (6052 ± 1307 ng/ml vs. 2934 ± 177 ng/ml; p = 0.055); (Fig. 2A). The serum levels of IL-10 were significantly lower in the RTX-treated rats compared with vehicle-treated controls.
(114 ± 12 pg/ml vs. 197 ± 12 pg/ml; p < 0.001; Fig. 2B). The values for TGF-1β did not differ between the groups (50.1 ± 1.1 ng/ml vs. 52.8 ± 1.2 ng/ml, p = 0.12).
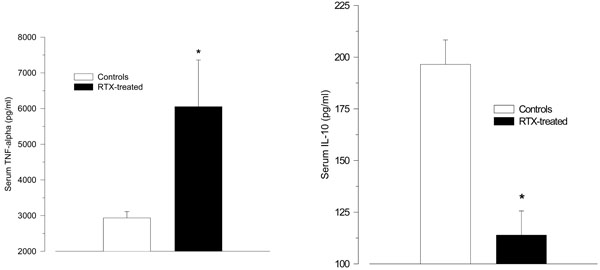
A-B. Serum levels of the pro-inflammatory cytokine tumour necrosis factor (TNF)-α (A), and the anti-inflammatory cytokine interleukin (IL)-10 (B) 2 h after intraperitoneal injection of LPS (150 µg/kg) in resiniferatoxin-treated rats and vehicle-treated control rats. Resiniferatoxin-treated rats tended towards higher serum levels of TNF-α (p = 0.06), but lower IL-10 levels (* p < 0.001 vs. controls).
Effects of Resiniferatoxin Pre-Treatment on Corticosterone Plasma Levels after LPS Challenge
Treatment with RTX significantly reduced the HPA axis response to LPS as measured by serum corticosterone (1504 ± 253 nmol/l vs. 2180 ± 41 nmol/l; p = 0.04; Fig. 3).
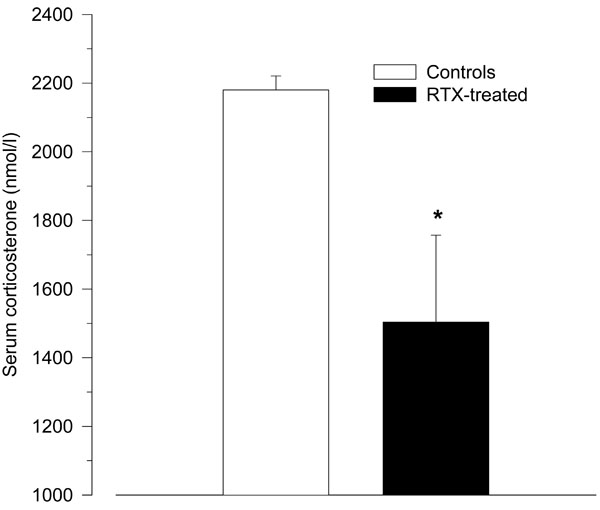
Serum corticosterone levels 2 h after intraperitoneal injection of LPS (150 µg/kg) in resiniferatoxin-treated rats and vehicle-treated control rats. Compared to control rats, resiniferatoxin-treated rats showed significantly lower serum levels of corticosterone (* p < 0.05 vs. controls).
DISCUSSION
The rationale for performing the present study was previous research showing that the reactivity of brain-controlled immunoregulatory systems plays a significant role in the susceptibility and resistance to periodontitis, principally due to the effects on immune responses [17-27, 32]. We have now expanded these results and shown that inhibition of sensory peptidergic neurons strengthens the resistance to ligature-induced periodontitis. The treatment also tended to boost the pro-inflammatory TNF-α serum levels after a robust in vivo LPS challenge, while at the same time inhibiting the anti-inflammatory cytokine IL-10 and the HPA axis-derived hormone corticosterone. The results support other recent in vivo studies demonstrating a pro-inflammatory role of sensory peptidergic neuron inhibition [14, 33, 34]. Furthermore, they suggest that the immune system influences the vulnerability to periodontitis in such a way that a stronger systemic TNF-α and a weaker HPA axis response ameliorates the clinical course of periodontitis [17-26]. To our knowledge, this is the first report showing that the autonomic sensory peptidergic nervous system, via the mechanisms outlined above, may be involved in the pathogenesis of periodontitis.
Increased colonization or over-growth of pathogenic microorganisms in the subgingival dental biofilm, including the Gram-negative Porphyromonas gingivalis, is commonly held to be responsible for initiating periodontitis [35]. This may indicate that adaptive immune system responses vital for clearing these pathogens are inappropriately regulated. It is also supported by our previous experimental studies in rodents showing that modulation of adaptive immune responses with agents that drive T helper 1 (Th1) and T regulatory (Treg) responses inhibits periodontitis [36, 37]. Other investigations may indicate that immune responses are dysregulated in patients with severe periodontitis [38-40]. The tissue injury itself seems to be caused by excessive recruitment and activation of cells belonging to the innate immune system, in particular polymorphonuclear phagocytes (PMNs), and subsequent release of tissue destructive mediators like reactive oxygen species and proteolytic enzymes [41-47].
Based on data from previous experiments, we have pointed out that periodontitis may be the result of a dysregulated brain-neuro-endocrine balance that reduces the ability of adaptive immunity to clear colonizing periodontopathogens. A chronic over-activity of the innate immune system may protect the gingival tissues as well as the entire organism from infection by these pathogens [25-27]. Thus, according to our hypothesis, periodontitis is not a result of failure to remove PMNs and their products from the inflammatory sites, as stated by other investigators [45], but rather a hidden benefit that protects the organism from infection of pathogenic dental plaque microorganisms during an inappropriate adaptive immune response.
It is well known that brain-controlled neurotransmitters and neuropeptides released from nerve terminals participate in local and systemic immuno-inflammatory reactions to infection or trauma [2, 7, 14]. Capsaicin and RTX have the unique property to excite or stimulate a subset of the sensory/peptidergic neurons when given in low doses, but to inhibit or block their function when given in repeated or high doses [15, 28, 29]. This desensitization of sensory peptidergic nerves may be caused by interaction with the TRPV1 receptor, a non-selective cation channel that is typically activated by noxious heat, acid or low pH, certain lipids, chemical irritants, and inflammatory mediators [28, 48]. TLR4 is co-expressed with TRPV1 on sensory peptidergic nerve terminals [16]. Thus, sensory peptidergic neurons may be able to respond both to inflammatory mediators and directly to constituents like LPS of Gram-negative bacteria. Pre-treatment with high and/or repeated doses of capsaicin or RTX eliminates this property, and local release of neuropeptides as well as afferent signals to the brain are discontinued [28, 48]. While the former should reduce inflammation, the latter would be expected to have the opposite effect. The net result of desensitization on the inflammatory response could therefore lean in either direction.
Originally, the neuropeptides from the sensory peptidergic nerve endings have been looked upon as primarily pro-inflammatory, but recent research has shown that they vary in their profile of action. Substance P is known to be a potent vasodilator, increasing vascular permeability and stimulating many pro-inflammatory processes, including LPS-induced TNF-α production and release [49, 50]. Calcitonin gene-related peptide and somatostatin, on the other hand, inhibit TNF-α [51-53]. In our in vivo study, the RTX-treated animals reacted towards a stronger TNF-α response to systemic LPS stimulation. We did not measure cytokine responses in gingival connective tissues in this experiment, but other investigations have also shown more powerful local inflammatory reactions and pro-inflammatory cytokine responses to LPS stimulation after RTX treatment [14]. This indicates that anti-inflammatory rather than pro-inflammatory pathways are inhibited by the RTX-induced inhibition of sensory peptidergic neurons. In line with this, it has recently been demonstrated that the TRPV1 agonist SA13353 inhibits LPS-induced TNF-α production in vivo [34]. An alternative explanation for the observed cytokine pattern implies that RTX may selectively inhibit the release of anti-inflammatory neuropeptides. The anti-inflammatory effect of calcitonin gene-related peptide and/or somatostatin release thus should outweigh the pro-inflammatory effect of Substance P release. Results from other investigators may be taken in favour of such an interpretation [14, 34].
The present experiments may also suggest that over-activation of the sensory peptidergic nervous system could cause a converse outcome. Interestingly, severe anxiety and major depression of the melancholic type has been found to increase the susceptibility to periodontitis in both humans and animal studies [26, 32, 54, 55], and chronic hyperactivity of the HPA axis and the sensory peptidergic nervous system is also a typical feature of these emotional conditions [3, 56, 57]. It is also of interest to note that non-steroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase 2 (COX- 2) inhibitors (Coxibs) inhibit periodontitis [58], and both classes of drugs suppress pain by inhibiting prostaglandin metabolites to activate the recently identified excitatory ion channel, transient receptor potential A1 (TRPA1) [59]. Incidentally, this receptor is co-expressed with the capsaicin receptor TRPV1 on primary sensory peptidergic neurons [59]. In addition, prostagandins, like pro-inflammatory cytokines and LPS, stimulate the HPA axis [60]. Thus, the protective effect of NSAIDs and Coxibs may in part be a result of reduced ability of the sensory peptidergic nervous system to signal and stimulate efferent brain controlled anti-inflammatory pathways, including the HPA axis. Based on this, one might speculate that chronic pain, e.g. in patients with pro-inflammatory autoimmune diseases like rheumatoid arthritis, may be a risk factor for developing severe periodontitis. Differences in pain experience may help us to understand why some investigators have found a negative [61], while others have found a positive association between periodontitis and rheumatoid arthritis [62]. Experimental animal studies as well as epidemiological studies are therefore needed to investigate the consequences of these putative risk factors.
CONCLUSION
Taken together, our principal finding is that inhibition or complete suppression of the sensory peptidergic nervous system with RTX significantly increases the resistance to ligature-induced periodontitis. The treatment also tended to promote a shift towards a stronger pro-inflammatory TNF-α, and a significantly weaker anti-inflammatory IL-10 and HPA-axis response to an in vivo challenge with LPS. Together with our previous research these data may help us to understand how differences in the reactivity of neural and hormonal pathways may influence the susceptibility to periodontitis.
CONFLICT OF INTEREST
The authors report no financial relationship related to any products involved in this study and declare that no funding has been available other than from the Norwegian Defence Research Establishment, Division of Protection, Kjeller, Norway.

