All published articles of this journal are available on ScienceDirect.

Metagenomic Analysis of Subgingival Microbiota Following Non-Surgical Periodontal Therapy: a Pilot Study
Authors Info & Affiliations
Abstract
This study tested the feasibility of a high throughput metagenomic approach to analyze the pre- and posttreatment of subgingival plaque in two subjects with aggressive periodontitis. DNA was extracted from subgingival samples and subjected to PCR amplification of the c2-c4 regions of the 16S rDNA using primers with bar codes to identify individual samples. The PCR products were pooled and sequenced for the v4 region of the 16S rDNA using the 454 FLX standard platform. The results were analyzed for species/phylotypes in the Human Oral Microbiome Database (HOMD) and Ribosomal Database Project (RDP) database. The sequencing of the amplicons resulted in 24,673 reads and identified 208 species/phylotypes. Of those, 129 species/phylotypes were identified in both patients but their proportions varied. While >120 species/phylotypes were identified in all samples, 28-42 species/phylotypes cumulatively represent 90% of all subgingival bacteria in each sample. The remaining species/phylotypes each constituted ≤0.2% of the total subgingival bacteria. In conclusion, the subgingival microbiota are characterized by high species richness dominated by a few species/ phylotypes. The microbiota changed after periodontal therapy. High throughput metagenomic analysis is applicable to assess the complexity and changes of the subgingival microbiota.
INTRODUCTION
Aggressive periodontitis representrs a distinct category of periodontal disease [1]. Localized aggressive periodontitis (LAP), formerly call localized juvenile periodontitis, is characterized by circumpubertal disease onset and destruction limited to central incisors and first molars. The clinical presentation of LAP is the strikingly normal appearance of gingival tissue, and the relatively low amounts of dental plaque and calculus. The major etiologic agent of LAP is Gram negative facultative Aggregatibacter actinomycetemcomitans. The generalize aggressive periodontitis is characterized by the relatively young ages of the affected individual (usually 30 years old or younger), and the extensive amounts of periodontal destruction in incisors, first molars and other permanent teeth.
It is now well recognized that periodontitis is a polymicrobial infection. However, only about half of the subgingival bacterial species or phylotypes are cultivable, which presents an obstacle to fully understand the causal relationship between subgingival bacteria and periodontitis [2]. Metagenomics involves the identification, sequencing, and functional and transcriptome analyses of environmental samples. The members of the microbial community are typically determined by tracking phylogenetic markers such as the 16S rDNA. Studies evaluating the subgingival bacterial diversity using a metagenomic approach were first undertaken by Kroes et al. [3] and Paster et al. [4]. Both studies employed a technique of PCR amplification of 16S rRNA genes of DNA from subgingival plaque to establish clone libraries of the amplicons for sequencing analysis. These two publications have spawned numerous studies using a similar strategy. Cumulatively, over 700 phylotypes of bacteria have been identified in the human oral cavity by cultivation and metagenomic approaches, with ~400 phylotypes detected in gingival crevices]. A few species have been implicated as the etiologic agents of periodontitis due to their strong association with the disease, while other species are thought to be beneficial to health [2-5]. Most of our understanding of subgingival bacteria comes from cultivable bacteria, however, and the role of many non-cultivable bacteria remains largely unknown.
Most of the studies by metagenomics examined the subgingival microbiota at single time points. There is limited information for the microbial changes following treatment. The objective of this study is to demonstrate the feasibility of metagenomic analysis for subgingival microbial changes after periodontal therapy.
MATERIALS AND METHODOLOGY
Study Subjects
The study was approved by IRB of University of Southern California. Two subjects (designated as D114 and D119) with generalized aggressive periodontitis were recruited from the Advanced Periodontology Clinic of the Herman Ostrow School of Dentistry of the University of Southern California. The Health Sciences Institutional Review Board of the University of Southern California approved the protocol. Both subjects signed an informed consent prior to their enrollment in the study. A diagnosis of aggressive periodontitis was determined based on the American Academy of Periodontology parameters [6]. The subjects did not have any systemic conditions that might contribute to their periodontal condition (i.e. uncontrolled diabetes); they had not taken antibiotics within the past year for any reason, nor had they received any non-surgical or surgical periodontal therapy in the past year.
Clinical Examination and Treatment
A single clinician measured clinical periodontal parameters, delivered treatment and obtained subgingival plaque samples. Clinical parameters measured were probing depth (PD), bleeding on probing (BOP), gingival recession (GR) and attachment level (CAL) on six sites per tooth for all remaining teeth. Both patients received Amoxicillin (500 mg) and Metronidazole (500 mg) TID for 8 days in conjunction with full mouth scaling and root planning completed over 2 visits.
Microbial Sampling
Subgingival plaque samples were obtained at baseline and at the 8-week follow-up so that a total of 2 samples before treatment and 2 samples after treatment were analyzed. Samples were taken from both maxillary first molars each with initial probing depths ≥5 mm. Sampling sites were isolated using cotton rolls after all supragingival plaque and calculus were removed using sterile Gracey curettes. Teeth were air dried and a total of 5 paper points were inserted into the depth of the pocket for 30 seconds. The paper points were then packed into one microcentrifuge tube per patient containing 1.5 mL phosphate buffer solution (PBS). The samples were vortexed to release the bacteria and stored at -70°C until ready for processing.
16S rDNA Based PCR Amplification & 454 Pyrosequencing
DNA from plaque samples were isolated and purified using the Qiamp DNA Mini-kit (Qiagen) according to the manufacturer’s instructions. Briefly, bacteria were pelleted by centrifugation and then suspended in Buffer ATL (all buffers provided by the manufacturer). Twenty microliters of Proteinase K were added, and the mixture was vortexed and incubated at 56°C on a rocking platform in a water bath. Buffer AL was added, vortexed and incubated at 70°C for 10 minutes. Ethanol was then added, vortexed and again centrifuged. The mixture was added to a QIAamp mini-spin column and centrifuged for 1 minute and the eluate was discarded. The column was then transferred into a 2 mL collection tube. After performing sequential rinses with Buffer AW1 and Buffer AW2, the column was placed into a new 2 mL collection tube and the eluent Buffer AE was added. The final eluate was collected and stored at -70°C. The concentration was determined by UV spectrometer (abs 260nm) and purity was determined by the A260/A280 ratio.
PCR primers were designed to anneal to the conserved C2 and C4 regions so that products would include the v3 and v4 regions (courtesy of Dr. David Fredricks, University of Washington). The forward primers comprised of a mixture 3 oligonucleotides (at a ratio of 3:1:1) with the following sequences (5’-3’) (i) GCCTCCCTCGCGCCATCAGGCACTCCTRCGGGAGGCAGCAG, (ii) GCCTCCCTCGC-GCCATCAGGCACACCTACGGGTGGCTGC, (iii) GCCT-CCCTCGCGCCATCAGGCACACCTACGGGTGGCAGC. The generic sequence of the reverse primers was GCCTTGCCAGCCCGCTCAG-(barcode)-GGACTACCV-GGGTATCTAAT. Four barcodes (AACCAACC, AACCAAGG, AACCATCG, AACCATGC) were used for the reverse primers to tag the 4 samples. The PCR amplification was performed in a total volume of 50 µl that included 50 ng DNA template, 0.2 µM forward primer mix, 0.4 µM reverse primer, 2.5 unit of PFUTurbo hotstart DNA polymerase (Strategene), 0.2 mM dNTPs and 1x reaction buffer. The PCR cycler was set so that 30 cycles of melting at 95°C for 30 seconds, annealing at 55°C for 40 seconds and extension at 72°C for 1 minute were performed. The final extension was at 72°C for 10 minutes. The amplification products were purified with QIAamp DNA mini kit (Qiagen, Valencia, CA), pooled and run on ¼ of a plate on the 454 sequencing FLX standard platform. The sequencing was performed from one end of the amplicons for the hypervariable v4 region of the 16S rDNA.
Sequence Analysis
Sequencing data were analyzed using the LibCompare and Pyrosequencing pipelines from the Ribosomal Database Project (RDP) website (http://rdp.cme.msu.edu). Sequences were sorted and designated according to the respective barcodes assigned to each sample. A classifier checked for the proper orientation of the sequence and possible chimeras. Assignments were made above 80% confidence threshold and results were listed in hierarchy views that showed the summary of the assignments and their respective significance values. Post processing was performed to assign sequences to the species level when possible. These results were then crosschecked with the NIH-supported Human Oral Microbiome Database (HOMD) project (http://www.homd.org).
Quantitative Real-Time PCR (qRT-PCR)
The amounts of A. actinomycetemcomitans, P. gingivalis and total bacteria in the samples were determined by qRT-PCR using an iCycler. The primers were designed to amplify the 16S rDNA of the following bacteria based on previously published studies [7, 8]: (i) A. actinomycetemcomitans, 5’-CTAGGTATTGCGAAACAATTTG-3’ and 5’-CCTGAAA-TTAAGCTGGTAATC-3’ (ii) P. gingivalis, 5’-AGGCAGC-TTGCCATACTGCG-3’ and 5’-ACTGTTAGCAACTACC-GATGT-3’, and (iii) universal primers for all bacteria: 5’-GATTAGATACCCTGGTAGTCCAC-3’ and 5’-TACCTT-GTTACGACTT-3’. Each PCR amplification was performed in a total volume of 25 µl containing 2 µl sample, 2.5 µl of each primers (3 µM), and 12.5 µl 2X Bio-Rad iQ SYBR Green supermix for real-time PCR. The thermocycler profiles were (i) 95°C/3 min, followed by 40 cycles of (95°C/15 sec, 55°C/30 sec and 72°C/30 sec) for A. actinomycetemcomitans, (ii) 95°C/10 minutes, followed by 45 cycles of (95°C/30 sec, 60°C/5 sec and 72°C/17 sec for P. gingivalis, or (iii) 95°C/10 min, followed by 45 cycles of (95°C, 52°C/5 sec and 72°C/30 sec) for all bacteria (universal primers). A melting curve analysis was performed to determine the specificity of the PCR products. The genomic DNA of P. gingivalis ATCC 33277 and A. actinomycetemcomitans D7S-1 were used to generate the respective standard curves for quantification. The genomic DNA of P. gingivalis ATCC33277 was used as template for PCR with the universal primers for quantification of total bacteria. Linear regressions were obtained by plotting the Log10 (ng input reference DNA) against the CT (cycle threshold) value. The slope of each standard curve was used to determine the reaction efficiency by the following equation: efficiency=10-1/slope - 1. All samples were run in triplicate to confirm the reproducibility of the data. CT values were then averaged for each triplicate to derive the amounts of specific DNA in the samples.
RESULTS
Clinical Observations
A summary of the clinical changes in both patients is presented in Fig. (1). There were reductions in the frequency of sites with bleeding on probing, and conversion of sites with probing depth of ≥6 mm to sites with probing depths of 4-5 mm. No statistical analysis was performed due to the small sample size.

Clinical changes after non-surgical periodontal therapy. All periodontal sites were examined for bleeding on probing (BOP) and probing depth (PD) for patients D114 (a) and D119 (b) after non-surgical periodontal therapy. Both patients received Amoxicillin (500 mg) and Metronidazole (500 mg) TID for 8 days in conjunction with full mouth scaling and root planning completed over 2 visits. The follow up clinical examinations were conducted 8 weeks after the baseline examination. There were reduction of BOP and PD (from ≥6 mm to PD of 4-5 mm) in both patients.
Pyrosequencing
The sequencing of the amplicons resulted in 24,673 reads with an average length of 252 base pairs. Among these reads 22,852 reads were of sufficient quality for species/phylotypes identification, and included 5,820 (122 species/phylotypes) and 5,946 (133 species/phylotypes) reads from patient D114 before and after treatment, and 5,994 (133 species/phylotypes) and 5,092 (100 species/phylotypes) reads from patient D119 before and after treatment. In total the analysis identified 208 species/phylotypes; 168 were found in D114 and 171 in D119. Of those, both patients presented with the same 129 species. Most of the species/phylotypes were detected in both patients (before or after treatment) but their proportions varied between patients (Fig. 2; more details in Table 1).
The % Composition of Species/Phylotypes in Subgingival Microbiota before and after Treatment
| Patient D114 | % Before | % After | Log2fold* | Patient D119 | % Before | % After | Log2fold* |
|---|---|---|---|---|---|---|---|
| F. nucleatum ss. nucleatum | 17.92% | 6.54% | -1.454 | F. nucleatum ss. nucleatum | 6.96% | 15.59% | 1.164 |
| P. gingivalis | 11.92% | 5.30% | -1.170 | Prevotella sp. | 2.07% | 12.57% | 2.603 |
| Prevotella sp. | 6.49% | 5.77% | -0.171 | Porphyromonas gingivalis | 13.56% | 0.12% | -6.847 |
| P. pseudoalcaligenes | 4.59% | 5.60% | 0.288 | A. actinomycetemcomitans | 10.58% | 2.34% | -2.178 |
| Prevotella intermedia | 5.43% | 4.54% | -0.258 | Prevotella oris | 0.87% | 9.68% | 3.480 |
| Streptococcus oralis | 0.33% | 9.40% | 4.848 | Treponema sp. | 7.39% | 0.00% | -11.878 |
| Synergistetes [G-3] sp. | 6.08% | 1.31% | -2.213 | Treponema denticola | 7.02% | 0.00% | -11.804 |
| Parvimonas micra | 2.49% | 2.03% | -0.292 | Veillonella atypica | 0.25% | 5.05% | 4.334 |
| Porphyromonas sp. | 2.51% | 1.82% | -0.466 | Porphyromonas sp. | 1.57% | 3.42% | 1.124 |
| Filifactor alocis | 2.49% | 1.80% | -0.469 | Leptotrichia sp. | 0.47% | 4.28% | 3.196 |
| Leptotrichia sp. | 1.22% | 2.99% | 1.295 | F. nucleatum ss. animalis | 1.47% | 3.10% | 1.080 |
| F. nucleatum ss. animalis | 1.20% | 2.49% | 1.049 | Filifactor alocis | 4.24% | 0.00% | -11.075 |
| Prevotella dentalis | 1.53% | 2.15% | 0.493 | Veillonella dispar | 0.13% | 3.97% | 4.893 |
| Fusobacterium naviforme | 3.52% | 0.12% | -4.903 | Synergistetes [G-3] sp. | 4.04% | 0.00% | -11.006 |
| Desulfovibrio fairfieldensis | 2.10% | 1.43% | -0.552 | Leptotrichia hofstadii | 0.12% | 3.59% | 4.944 |
| Streptococcus sinensis | 0.12% | 3.35% | 4.798 | Bacteroidetes [G-2] sp. | 3.09% | 0.00% | -10.618 |
| Actinomyces sp. | 0.19% | 3.13% | 4.049 | Corynebacterium matruchotii | 0.37% | 2.67% | 2.863 |
| Rothia dentiocariosa | 0.00% | 3.25% | 10.883 | Prevotella oulorum | 0.00% | 2.99% | 10.805 |
| Treponema sp. | 2.58% | 0.64% | -2.012 | TM7 [G-1] sp. | 0.28% | 2.53% | 3.159 |
| F. nucleatum ss vincentii | 2.37% | 0.15% | -3.969 | T. forsythia | 2.72% | 0.00% | -10.435 |
| K. oralis | 0.00% | 1.98% | 10.174 | Tannerella sp. | 0.02% | 2.26% | 7.081 |
| Cardiobacterium hominis | 0.05% | 1.88% | 5.191 | Treponema medium | 2.10% | 0.06% | -5.157 |
| Aggregatibacter paraphrophilus | 0.07% | 1.83% | 4.737 | Bacteroidetes [G-5] sp. | 2.12% | 0.00% | -10.075 |
| Streptococcus peroris | 0.03% | 1.78% | 5.697 | Desulfobulbus sp. | 2.04% | 0.00% | -10.017 |
| Rothia mucilaginosa | 0.05% | 1.72% | 5.057 | Desulfovibrio fairfieldensis | 2.02% | 0.00% | -10.006 |
| Dialister invisus | 1.62% | 0.02% | -6.585 | P. pseudoalcaligenes | 1.58% | 0.16% | -3.335 |
| Fusobacterium periodonticum | 0.00% | 1.60% | 9.861 | Bacteroidetes [G-1](Prevotella) sp. | 1.57% | 0.00% | -9.641 |
* For non-detectable bacterial species (0%) 1 sequence read was used to calculate fold-change
Comparison of the Subgingival A. Actinomycetemcomitans and P. Gingivalis Determined by qRT-PCR vs. by Metagenomic Analysis
| Species | Patient D114 | Patient D119 | ||
|---|---|---|---|---|
| Before Treatment | After Treatment | Before Treatment | After Treatment | |
|
% A. actinomycetemcomitans Real-time PCR Metagenomic analysis |
0.01 0 |
0.02 0.25 |
1.32 10.58 |
0.63 2.34 |
|
% P. gingivalis Real-time PCR Metagenomic analysis |
23.27
11.92 |
1.88
5.30 |
11.19
13.6 |
0.01
0.12 |
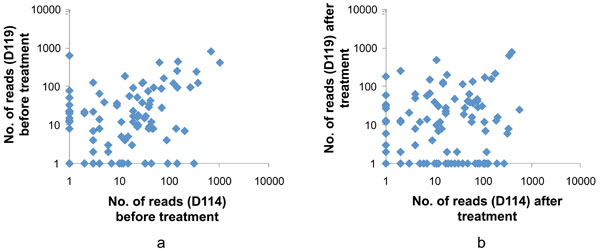
Comparison of the subgingival microbiota in subjects D114 and D119 before (a) and after (b) nonsurgical periodontal treatment. Each dot represents the number of a specific sequencing read (matched to a species/phylotype) identified in the subgingival samples. A total of 121 species/phylotypes were shown in the analysis after data were filtered to include only reads that reached a count of >10 in at least one of the 4 samples. Most of the species/phylotypes were detected in both patients (before or after treatment) but their proportions varied between patients.
While >120 species/phylotypes were identified in all samples, 85-90% of the species/phylotypes were represented by ≤100 sequencing reads (≤0.2% of the total reads). The unevenness (i.e., domination by a few species/phylotypes) of the subgingival bacterial community is illustrated by the cumulative % of total reads by species/phylotypes in Fig. (3). 50% and 90% of the total reads in subgingival plaque were accounted for by 6 and 28 species/phylotypes in samples from D114 before treatment, 11 and 42 species/phylotypes in sample from D114 after treatment, 6 and 36 species/phylotypes in D119 before treatment, and 6 and 32 species/phylotypes in D119 after treatment.
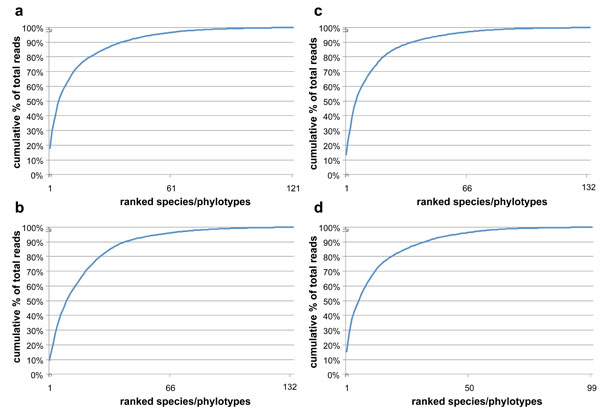
Cumulative % of total reads with each addition of ranked species/phylotypes from the highest to the lowest read counts. (a) & (b), D114 before and after treatment; (c) & (d), D119 before and after treatment. The subgingival microbiota were diverse but dominated by relatively few species.
Changes in the Subgingival Microbiota after Nonsurgical Periodontal Treatment
Fig. (4) shows the log2(fold change) for the most commonly found 121 species/phylotypes in samples after treatment. While significant microbial changes occurred after periodontal therapy, there were no consistent patterns in the levels of changes between subjects. A more detailed analysis of the changes for the 27 most frequent species before and after periodontal treatment is presented in Table 1. In general, half of the species/phylotypes increased while the other half decreased by more than 2-fold after treatment. Red complex bacterial species (P. gingivalis, T. denticola and T. forsythia) were among the putative pathogenic species that showed the greatest decrease after treatment. In contrast, several Gram-positive Streptococcus, Rothia, Actinomyces and the Gram-negative Veillonella were among the bacteria that showed the greatest increases after treatment.
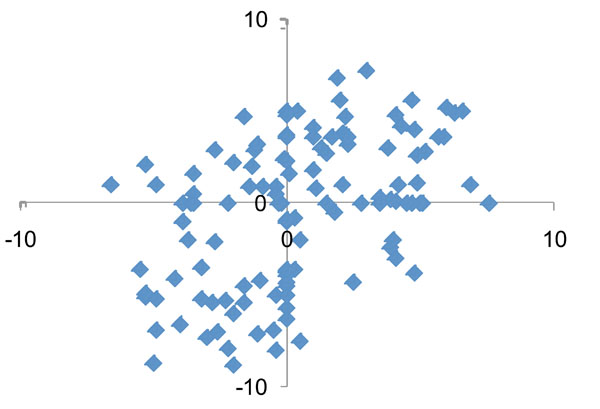
Comparison of quantitative changes in the subgingival microbiota after nonsurgical periodontal therapy between the two study patients. Each dot represents the log2(fold change following treatment) of a species/phylotype in D114 (x-axis) and D119 (y-axis). No correlation of the microbial changes was detected between the two patients.
The % of subgingival A. actinomycetemcomitans and & P. gingivalis determined by qRT-PCR are shown in Table 2.
The levels and the proportions of P. gingivalis in subgingival plaque before and after treatment were similar to those calculated from sequencing reads in both patients. The low levels of A. actinomycetemcomitans in subgingival plaque of patient D114 before and after treatment were confirmed by both quantification methods. In the samples from patient D119, lower levels of subgingival A. actinomycetemcomitans were found based on real time PCR than by metagenomic analysis. However, the trends for reduction of the bacterium after treatment were verified by both quantification protocols.
DISCUSSION
The pilot study was designed to evaluate metagenomics as an approach to monitor subgingival microbiota. While only two subjects were involved (with each subject contributing one pooled sample from two periodontal sites before and after treatment), the data were informative and had provided a guideline for future studies. We have achieved sufficient sequencing depths to account for any bacterial species that constituted 0.02% or greater of the subgingival microbiota. Increasing the sequencing depths may not be necessary. Using barcoded primers to tag amplified DNA samples, it is feasible to sequence approximately 100 samples on a single 454 FLX standard platform run. With further reduction in sequencing costs and better sequencing technology, the metagenomic approach will be applicable for large scale clinical studies to evaluate the outcomes of periodontal therapy.
In this study, the subgingival microbiota were high in species richness but dominated by a few species/phylotypes, with significant shifts of microbiota that occurred after treatment. Also, the compositions of the subgingival microbiota were highly individualized. The observed species richness in the subgingival microbiota is in general agreement with other studies. Kroes et al. [3] examined two sites (teeth #3 and #30) of a single patient and estimated that a total of 84 phylotypes could be identified in the sites. The level of species richness is similar to this study in which approximately 100 to 130 species/phylotypes were identified in each sample. While there is a lack of information on the evenness of subgingival microbiota, our results are in general agreement with the previous estimates [3].
Pre- and post-therapy samples showed significant quantitative and distributive species changes. Quantitatively, many known pathogenic species went from easily detectable to zero counts while the opposite occurred for many known health-associated bacteria. In almost all cases, the counts of these known pathogens decreased significantly, with some species, such as P. gingivalis, T. forsythia, T. denticola, and P. micra decreasing to almost zero counts after treatment. Conversely, known commensals such as S. sanguinis and K. oralis [9] increased after treatment, ranging from 2-fold increases up to 100-fold increases. Other suspected periodontopathogens, such as those found in greater abundance in refractory periodontitis patients by Colombo et al. [10] and periodontally diseased patients by Paster et al. [4], including Fusobacterium naviforme, Dialister invisus, Filifactor alocis, Eubacterium saphenum, and Desulfobulbus sp. were also found to have significantly decreased in both patients after treatment. The potentially pathogenic species Desulfovibrio fairfieldensis observed by Loubonix et al. [11] decreased appreciably in both patients while the suspected pathogen observed by Paster et al. [4], Fusobacterium nucleatum ss. animalis, surprisingly increased in both patients. Likewise, other suspected pathogens, Selenomonas infelix and Bacteroides zoogleoformans [12] increased after treatment. Of the more recently suspected commensals, Cardiobacterium hominis, Capnocytophaga sputigena, Campylobacter showae, Corynebacterium matruchotii, and Rothia muclaginosa [10,13] all increased, on average, 30-fold in patient D114.
The assignments of bacterial species to either pathogen or commensal have not always been consistent in the literature. Aas et al. [13] found Gemella sanguinis to be part of the normal subgingival flora of healthy subjects while Colombo et al. [10] noted it as being more prevalent in refractory periodontitis patients. Similarly, Campylobacter gracilis and Granulicatella adiacens were deemed pathogenic by Colombo et al. [10], while Aas et al. [13] described them as some of the most predominant subgingival species found in health. Rothia dentocariosa was described by both Aas et al. [13] and Colombo et al. [10] as being more predominant in health while Paster et al. [4] noted it more commonly in disease. In patient D114, counts of Gemella sanguinis, Granulicatella adiacens, and Rothia dentiocariosa significantly increased after treatment, as did Camplyobacter gracilis in patient D119, indicating that, as clinical parameters improved after treatment, perhaps these species may be considered more beneficial than pathogenic.
Of the more abundant but lesser known species detected in these samples, associations with other environments or diseases have been noted. Pseudomonas pseudoalcaligenes, the fourth most prevalent species both before and after treatment in patient D114, can be found in environmental literature in reference to cyanide and metalwork. Leptotrichia sp., present in significantly large numbers after treatment in both patients, has been frequently studied with regard to the cervicovaginal microflora [14]. Chloroflexi can be found most commonly in the literature regarding wastewater treatment sludge [15]. The Lachnospiraceae spp., which generally increased after treatment, have previously been associated with caries-free children [16].
Several authors have published universal primer sequences to exploit distinct conserved regions of the 16s rRNA gene. However, in a review of the 16s rRNA gene of E. coli, Baker et al. [17] noted that, in fact, few totally conserved sequences actually exist. Within a sample of 500 bases of E. coli, roughly 10% of the bases can be considered totally conserved amongst all species. Furthermore, continuous sequences of those totally conserved bases are typically less than 4 sequential bases. While this means that no primer of sufficient length could be designed as a “100% match universal primer,” the majority of conserved sequences that are commonly exploited show some variability between species, and evidence shows that a 70% identity is sufficient for successful amplification and identification of distinct species. Thus, while a universal primer has itself some innate variability, it is more likely that the sequence similarity in the conserved regions and sequence difference in the hypervariable regions is sufficient to identify and differentiate species.
qRT-PCR was employed to determine the levels of A. actinomycetemcomitans and P. gingivalis before and after treatment. The results for P. gingivalis were consistent between metagenomic analysis and qRT-PCR. On the contrary, the quantification of the subgingival A. actinomycetemcomitans was substantially different between these two methods. The discrepancies were likely due to biased amplification of the 16S rDNA in different bacterial species. Nevertheless, the proportions and the fold-changes after treatment for A. actinomycetemcomitans as assessed by qRT-PCR were still in the same direction and within one order of magnitude in comparison to quantification based on sequencing reads. The results suggested that quantification by sequencing reads may be a reasonable approach to assess the levels of subgingival bacterial species.
CONCLUSION
This study examined the subgingival species/phylotypes by high-throughput metagenomic analysis for two generalized aggressive periodontitis patients. The sequencing depths were sufficient to identify bacterial species that constituted more than 0.02% of the total subgingival bacteria. The approach was sufficient to reveal the richness and unevenness of the subginigval bacterial community, as well as the changes of the community following periodontal treatment. The approach can be scaled up for a larger set of subjects to examine the causal relationship between the subgingival microbiota and periodontal health and disease.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
This study was supported in part by NIDCR grant R01 DE12212.

