All published articles of this journal are available on ScienceDirect.

Odontohypophosphatasia Diagnosed through Isolated Dental Findings: A Longitudinal Case Report
Abstract
Background
Hypophosphatasia (HPP) is a rare inherited metabolic disorder of low prevalence, characterized by low activity of tissue-nonspecific alkaline phosphatase (TNSALP), causing defective mineralization of teeth and bone. Odontohypophosphatasia (odonto-HPP) is the mildest form of hypophosphatasia, with manifestations limited to dental features, such as premature loss of primary teeth.
Case Report
This case report presents a 3-year-old female patient who was diagnosed with odonto-HPP. There was a history of premature exfoliation of the lower primary central incisors shortly after eruption, without any accompaniment of pain or bleeding. She was referred at the hospital on a clinical suspicion of hypophosphatasia for further evaluation. The diagnosis of odonto-HPP was confirmed following bone densitometry and genetic analysis, which revealed a subtle mutation of the ALPL gene. Clinical assessment revealed normal body growth without systemic defects. Mandibular anterior region showed bone resorption and gingival recession, whereas the overall eruption pattern of teeth was normal. Treatment included vitamin D supplementation and dietary counseling. The patient is currently receiving follow-up every two years.
Conclusion
The case, as the second reported case of HPP on Lesvos Island, highlights the importance of recognizing early dental manifestations of the condition and the critical role of the dentist in the recognition and referral of such cases. Early diagnosis promotes effective surveillance and avoids unwarranted intervention.
1. INTRODUCTION
Hypophosphatasia (HPP), a rare inherited disorder, is identified by impaired mineralization in bone and teeth [1-3]. It is caused by the loss-of-function variant in the ALPL gene, located on chromosome 1p36.12, which encodes Tissue Non-specific Alkaline Phosphatase (TNSALP or TNAP) [4, 5]. More than 300 distinct mutations in the TNSALP gene have been reported as causes of HPP, across various ethnic groups and populations, exhibiting either an autosomal dominant or an autosomal recessive pattern [6]. The deficiency of TNSALP is typically indicated by a reduced serum total alkaline phosphatase (ALP) level. Serum ALP measurement is readily available in most clinical settings and serves as a sensitive screening tool for HPP when the clinical history aligns with the condition [7]. Given that physiological ALP levels are typically elevated during childhood, it is essential to use an age- and gender-specific reference range when interpreting results [8]. Hypophosphatasia presents with a broad clinical spectrum, from the often lethal perinatal type to the mild adult form without pathognomonic symptoms [2, 9,-12].
HPP is traditionally classified by age of onset and presence of bone disease [7]. The main subtypes include the perinatal form, with onset in utero and typically lethal, the infantile form, with onset before 6 months of age and a mortality rate of approximately 50% due to respiratory failure, the childhood form, with onset between the ages of 6 months and 18 years, and the adult form, with on-set occurring after the age of 18 [7, 10, 13]. It is important to note that other conditions may also lead to low ALP levels, including hypothyroidism [13], multiple myeloma, Cushing's syndrome, severe anemia, and vitamin D intoxication [7, 13]. These conditions can be differentiated through clinical history and laboratory investigations. TNSALP deficiency leads to elevated levels of specific substrates in urine and plasma, which can serve as diagnostic biomarkers. These include inorganic pyrophosphate (PPi), pyridoxal-5′-phosphate (PLP), the predominant circulating form of vitamin B6, and phosphoethanolamine [7, 14]. Increased extracellular concentrations of PPi impair bone mineralization by inhibiting the growth of hydroxyapatite crystals [15]. In severe cases, TNSALP is unable to sufficiently dephosphorylate PLP to pyridoxal, a process required for its passage across the blood-brain barrier and its role as a cofactor in neurotransmitter biosynthesis. This can result in vitamin B6-responsive seizures [16-18].
The sequelae of HPP may include muscle weakness, abnormal gait, bone deformities, osteomalacia, premature loss of teeth, and dental caries [7]. In more severe forms, additional complications may include failure to thrive, craniosynostosis, seizures, nephrocalcinosis, and respiratory compromise due to rachitic chest [9]. Odontohypophosphatasia (odonto-HPP) is the mildest and most common form, manifesting exclusively with dental complications at any age, without radiographic or histopathologic signs of rickets or osteomalacia [19]. Typical findings include premature loss of one or more deciduous teeth, often before the age of five, without associated pain or bleeding. The underlying cause is a deficiency of mineralized cementum, which impairs the attachment of the root to the periodontal ligament, resulting in the exfoliated root remaining intact [20]. Odonto-HPP is diagnosed when dental manifestations are the only clinical signs, accompanied by the biochemical hallmarks of HPP. Consequently, additional diagnostic procedures, such as radiological imaging and, if necessary, bone biopsies, should be performed to rule out rickets or osteomalacia [15, 19]. Common dental abnormalities include defective cementum and enamel formation, enlarged pulp chambers, and the premature loss of fully rooted primary teeth before the age of five, as well as early loss of permanent teeth [21, 22]. Adult-onset HPP is typically identified when skeletal symptoms appear during middle age; however, affected individuals often recall having experienced premature tooth loss during early childhood [13].
In this study, we present a case report of a pediatric patient in whom premature exfoliation of primary teeth was the initial and sole clinical manifestation of hypophosphatasia, highlighting the critical role of dental findings in the early diagnosis of the condition. This case report follows the CARE guidelines. Ethical review and approval were not required for this study, following current national guidelines and Greek legislation (Law No. 3328/2005). Written informed consent for publication was obtained from the patient’s legal guardian.
2. CASE REPORT
A 3-year-old female patient presented to a dental practice in Lesvos, Greece, in 2015, following the exfoliation of her lower primary central incisors shortly after their eruption. According to her parents, this was initially believed to be self-induced, prompting them to seek dental consultation. Clinical examination revealed intact roots on the exfoliated teeth, no gingival inflammation or bleeding, and no mobility of the other teeth. These findings were consistent with cementum deficiency rather than inflammatory periodontal disease. Anthropometric measurements were within the normal range for age and sex, and family history was negative for premature loss or metabolic disorders. Upper and lower occlusal radiographs were obtained to assess the status of her dentition (Fig. 1). Based on the clinical findings and history, hypophosphatasia (HPP) was suspected, and the child was referred to the hospital for diagnostic workup. Metabolic testing revealed markedly elevated plasma pyridoxal-5′-phosphate (PLP) levels, consistent with tissue-nonspecific alkaline phosphatase deficiency.
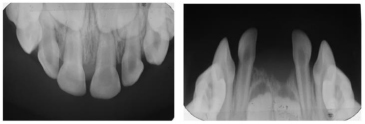
Upper and lower occlusal radiographs at age 3, following exfoliation of the mandibular primary central incisors, demonstrating a normal eruption pattern and absence of pathological changes.
The diagnosis of odonto-HPP, the mildest form of HPP, was confirmed through bone densitometry and genetic testing, which indicated a mild phenotype without skeletal manifestations. Bone densitometry showed a TBLH (BMD) of 0.746 g/cm3 (Z-score +0.2; 102% of the age, sex, and size-matched mean) with all regional Z-scores between -0.1 and +1.0, within the expected range for age. Genetic analysis by direct sequencing of all coding exons and flanking intronic regions of ALPL revealed a heterozygous mutation (c.550C>T, p.Arg184Trp), associated with mild phenotypes, such as odonto-HPP. A benign polymorphism (c.455G>A, p.Arg152His) was also identified. A daily 10 μg (400 IU) vitamin D supplement was administered for five months, with periodic cycles thereafter, as the sole medical intervention. Dietary guidance emphasized calcium intake, particularly dairy products, and regular physical activity. Follow-up evaluations were scheduled every two years at a pediatric hospital, including dental and orthopedic assessments, bone densitometry, blood tests, and general examinations. The overall prognosis was favorable.
At the 2021 follow-up, when the patient was 9 years old and in the mixed dentition stage, oral examination indicated moderate oral hygiene. Clinically, the eruption sequence of permanent teeth appeared age-appropriate. A panoramic radiograph confirmed these findings, showing localized vertical bone loss adjacent to the mandibular central incisors, while the remaining dentition displayed normal eruption patterns without other pathological changes (Fig. 2). Professional cleaning and scaling were performed. Preventive measures were reinforced, including topical fluoride application and placement of sealants on the permanent first molars, aiming to enhance caries resistance during the mixed dentition phase.
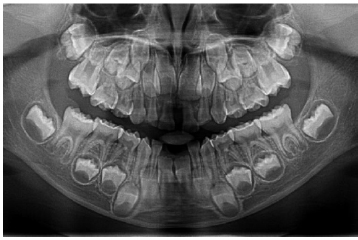
Panoramic radiograph at age 9 (2021), showing localized vertical alveolar bone loss adjacent to the mandibular central incisors, with the remaining dentition demonstrating a normal eruption sequence.
At the subsequent follow-up appointment in February 2023, when the patient was 11 years old, she returned for further evaluation. All diagnostic test results were within normal ranges, and routine systemic examinations were unremarkable. Clinically, she demonstrated normal physical development and age-appropriate dentition, with no systemic abnormalities. In the mandibular anterior region, localized labial bone resorption was noted around the permanent central incisors, accompanied by buccal gingival recession measuring approximately 3 mm. The gingival tissues appeared otherwise healthy, without bleeding or inflammation, and no abnormal mobility of the incisors was detected. Mild anterior crowding was evident in both maxillary and mandibular arches; however, no significant malocclusion, such as a crossbite or open bite, was observed. A panoramic radiograph confirmed the clinical findings, showing localized alveolar bone loss limited to the mandibular central incisors, while the remaining dentition followed a normal eruption sequence for the mixed dentition stage (Fig. 3).
Extraoral assessment revealed a symmetrical facial profile, competent lips, a normal smile line, and an orthognathic skeletal relationship. Intraoral examination showed healthy soft tissues, a normal tongue, and several composite restorations placed on posterior teeth. The overall oral condition was stable, with no new systemic or dental complications reported.
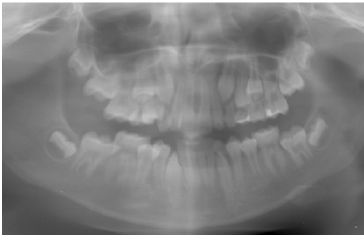
Panoramic radiograph at age 11 (2023), confirming localized alveolar bone resorption in the mandibular central incisors, with the remaining dentition showing a normal eruption.
3. DISCUSSION
The diagnosis of this case was initiated following the premature exfoliation of deciduous teeth in the absence of skeletal symptoms, underscoring the critical role of dental professionals in the early detection of systemic conditions. The mild phenotype and lack of skeletal involvement are associated with a favorable prognosis. Regular re-evaluation is warranted to detect any emerging changes. Notably, this is the second recorded case of HPP on the island of Lesvos, contributing valuable epidemiological insight. The pathogenicity of odonto-HPP is supported by the identification of the heterozygous c.550C>T (p.Arg184Trp) mutation, highlighting the utility of genetic testing in ambiguous cases. This case illustrates how the timely detection of early dental signs can enable prompt diagnosis, appropriate management, and the avoidance of unnecessary interventions in patients with rare metabolic disorders.
Premature exfoliation of primary teeth can be attributed to a wide range of etiologies, including environmental factors (acrodynia, autoextraction), genetic conditions (Ehlers-Danlos syndrome, Papillon-Lefevre syndrome), endocrine (hypophosphatemia, hypophosphatasia), or immunological conditions (prepubertal periodontitis, leukemia) [23]. In this case, these alternatives were excluded through clinical and laboratory evaluation. The exfoliated teeth showed intact roots without resorption; there was no evidence of gingival inflammation, bleeding, or tooth mobility suggestive of aggressive periodontitis, and the patient’s systemic evaluation was normal. The combination of premature exfoliation with intact roots, elevated PLP levels, and identification of a pathogenic ALPL mutation confirmed the diagnosis of odonto-HPP.
To date, only a limited number of reports have identified specific ALPL mutations associated with odonto-HPP [24-30]. These studies collectively demonstrate the genetic heterogeneity of odonto-HPP, with both missense and nonsense variants reported in patients of different populations. Jiang et al. [31] described odonto-HPP due to compound heterozygosity (c.346G>A and c.1563C>G), where a sibling with a single variant remained asymptomatic. This variability in penetrance parallels our patient with the heterozygous p.Arg184Trp variant.
In comparison to other studies, and in contrast to most patients described by Mori et al. [7], who received a delayed diagnosis of HPP only after developing musculoskeletal symptoms despite presenting early dental signs, this case emphasizes the importance of early recognition of dental manifestations, allowing timely referral by a pediatrician or a dentist. Although no extra-dental symptoms have been observed thus far, continued monitoring is necessary for the long-term well-being of the patient.
This case also substantiates the findings of Spodzieja and Olczak-Kowalczyk (2022) [32], who have identified hypophosphatasia to manifest solely with premature exfoliation of primary teeth due to cementum hypoplasia. HPP has been identified as a significant diagnostic consideration in cases of premature tooth loss without root resorption, caries, or trauma. The clinical presentation of the patient, i.e., absence of skeletal features and radiographic features suggestive of cementum deficiency, further highlights the importance of early dental evaluation and genetic investigation in such a situation.
A similar clinical presentation was reported by Hollis et al. in 2013 [33], involving a 20-month-old female with odonto-HPP, with premature exfoliation of lower primary central incisors (teeth 71 and 81). Radiographic assessment revealed extensive bone loss in the affected region, with no root resorption signs. Histological analysis revealed a complete absence of both cellular and acellular cementum. These findings are consistent with the present case, wherein gingival recession and alveolar bone resorption occurred in the mandibular anterior segment following premature loss of primary incisors. Bone changes in odonto-HPP can result in localized instability, underscoring the importance of continued clinical and radiographic follow-up in such individuals.
The presence of the c.550C>T (p.Arg184Trp) mutation in a heterozygous state has been associated in the literature with milder expressions of hypophosphatasia, like odonto-HPP. As noted by Mornet et al. [34], this specific variant frequently results in isolated dental manifestations, without involvement of the skeleton, particularly if not preceded by a second pathogenic allele. These findings are aligned with the case presented, which involved premature exfoliation of primary teeth without any systemic involvement.
In patients with perinatal or infantile HPP, enzyme replacement therapy (ERT) with asfotase alfa (Strensiq™; Alexion Pharmaceuticals, Inc., USA) has emerged as a recent treatment option [35-38]. This therapy improves respiratory and motor function, as well as skeletal symptoms, by increasing circulating levels of tissue-nonspecific alkaline phosphatase (TNSALP). Fracture healing is enhanced through reduced osteoid accumulation and improved mineralization of bone matrix, thereby decreasing fracture risk [39, 40]. The efficacy of ERT has shown promise across various age groups, including infants, children, adolescents, and even adults who have pediatric-onset disease [41].
Although studies in mouse models of pediatric HPP have demonstrated beneficial effects of ERT on dental tissues, corresponding evidence in humans remains limited and largely observational [42]. In mice, chimeric and bone-targeted alkaline phosphatase has been shown to partially restore the microstructure of alveolar bone, acellular cementum, and dentin. However, when administered from birth, it may interfere with enamel formation, resulting in decreased enamel mineralization and reduced thickness [43-46]. Clinical case reports have documented that asfotase alfa therapy may reduce tooth mobility and delay the premature exfoliation of deciduous teeth in HPP patients [29, 47]. Nevertheless, most studies investigating the effects of asfotase alfa lack detailed dental assessments, limiting conclusions about its efficacy in treating the dental-periodontal manifestations of HPP [48].
Although the current literature supports the present findings, additional case series and long-term follow-up data would be valuable to better define the full phenotypic spectrum of odonto-HPP.
A key strength of this case report lies in the early recognition of odonto-HPP based solely on dental findings, which enabled timely and accurate diagnosis through genetic testing. Additionally, this case contributes to the limited epidemiological data on hypophosphatasia cases in the specific geographical area, as the second reported case on the island of Lesvos.
4. LIMITATIONS
This single-patient case of odonto-HPP inherently limits the generalizability of its findings and precludes direct extrapolation to all affected individuals. The lack of a comparative cohort further restricts drawing broader conclusions about prognosis or management. Nonetheless, the rarity and exclusive dental presentation of odonto-HPP make this case valuable for clinical and epidemiological insight. Its longitudinal follow-up and genetic confirmation of a rare ALPL variant underscore the pivotal role of dental professionals in detecting rare metabolic disorders.
CONCLUSION
This case highlights the importance of recognizing early signs of odonto-HPP and underscores the pivotal role of dental professionals in identifying clinical features that may signal an underlying systemic disorder. The role of dentists is essential for initiating timely referrals, facilitating early diagnosis, and enabling appropriate multidisciplinary management. Early detection allows for patient-specific treatment planning, ongoing monitoring, and prevention of unnecessary interventions.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: N.K.: Study conception, design, and writing of the manuscript; N.K. and A.A.: Manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| HPP | = Hypophosphatasia |
| Odonto-HPP | = Odontohypophosphatasia |
ETHICAL STATEMENT
According to current national guidelines and Greek regulations (Greek legislation No. 3328/2005), ethical approval from an ethics committee is not necessary for single-patient case reports.
CONSENT FOR PUBLICATION
Written informed consent for publication of this paper was obtained from the patient’s legal guardian.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article will be available from the corresponding author [N.K] upon reasonable request.
ACKNOWLEDGEMENTS
Declared none.

